
The Genetics of Coronary Artery Disease: A Vascular Perspective

Abstract
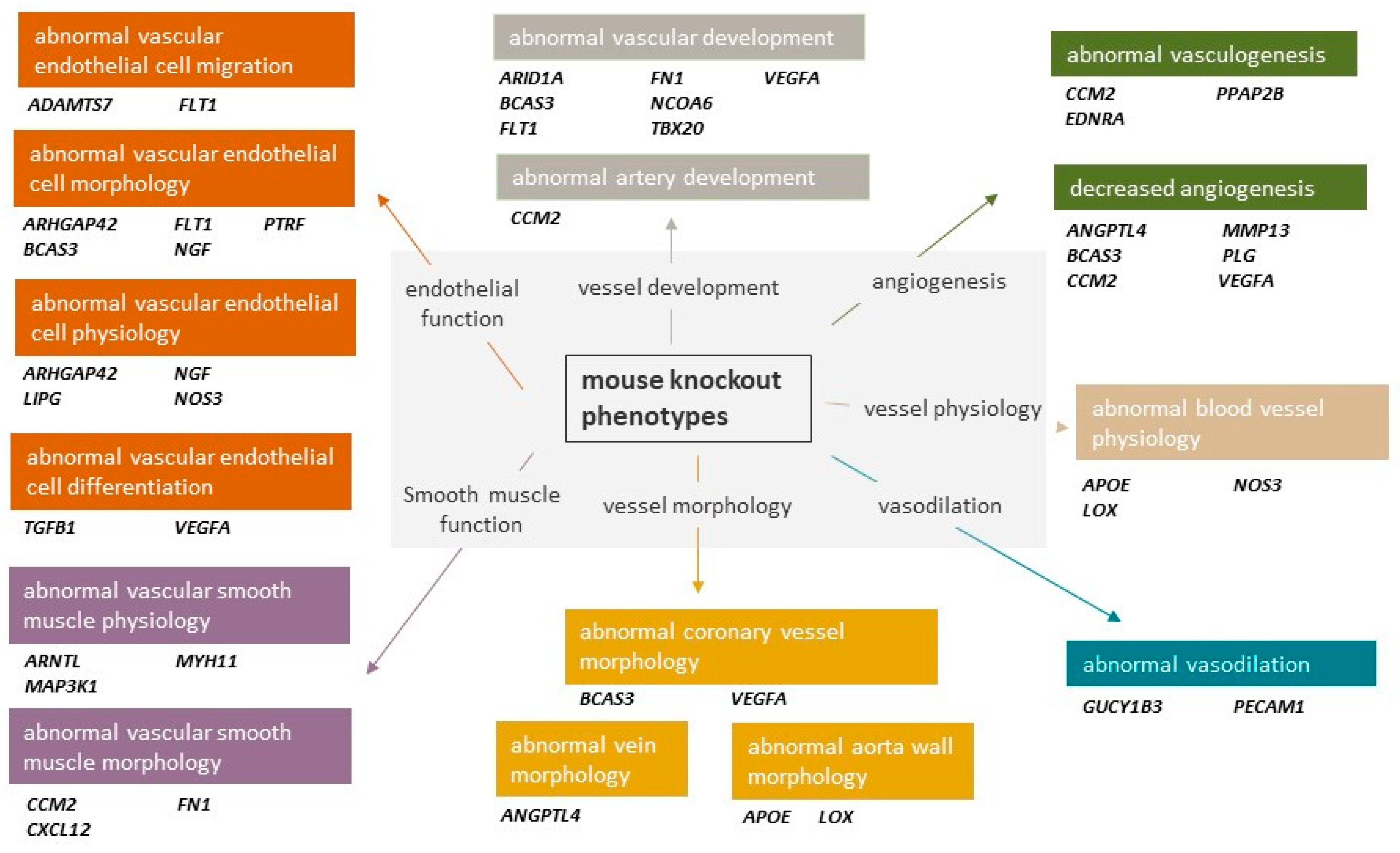
:1. Introduction
1.1. Vascular CAD GWAS Genes and Pathways
1.1.1. Vascular Endothelial Cell Dysfunction
JCAD
AIDA
PLPP3
MAT2A
DHX38
ARVCF
MYO9B
FES/FURIN
1.1.2. Vascular Smooth Muscle Cell Dysfunction
9p21 Locus
SMAD3 and TGFB1
ZEB2
TWIST1
MIA3
TCF21
PDGFD
MFGE8 and MAP3K11
1.1.3. Neovascularisation
VEGFA and FLT1
PTK7
BCAS3
Extracellular Matrix Remodelling
FN1
MMP13
2. Characterisation of Functional Vascular CAD Variants
2.1. Functionally Informed Fine-Mapping Studies
2.2. Expression Quantitative Trait Loci
2.3. Massively Parallel Reporter Assays
2.4. Genome Editing
Knock-Out (CRISPRko)
2.5. Transcriptional Modifications CRISPR
2.6. Single-Base CRISPR
3. Therapeutic Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Kathiresan, S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat. Rev. Genet. 2017, 18, 331–344. [Google Scholar] [CrossRef]
- Zdravkovic, S.; Wienke, A.; Pedersen, N.L.; Marenberg, M.E.; Yashin, A.I.; De Faire, U. Heritability of death from coronary heart disease: A 36-year follow-up of 20,966 Swedish twins. J. Intern. Med. 2002, 252, 247–254. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Claringbould, A.; Zaugg, J.B. Enhancers in disease: Molecular basis and emerging treatment strategies. Trends Mol. Med. 2021, 27, 1060–1073. [Google Scholar] [CrossRef]
- Gupta, R.M.; Schnitzler, G.R.; Fang, S.; Lee-Kim, V.S.; Barry, A. Multiomic Analysis and CRISPR Perturbation Screens Identify Endothelial Cell Programs and Novel Therapeutic Targets for Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 600–608. [Google Scholar] [CrossRef]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.-K.; Okada, Y.; et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020, 52, 669–679. [Google Scholar] [CrossRef]
- Aragam, K.G.; Jiang, T.; Goel, A.; Kanoni, S.; Wolford, B.N.; Atri, D.S.; Weeks, E.M.; Wang, M.; Hindy, G.; Zhou, W.; et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022, 54, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Tcheandjieu, C.; Zhu, X.; Hilliard, A.T.; Clarke, S.L.; Napolioni, V.; Ma, S.; Lee, K.M.; Fang, H.; Chen, F.; Lu, Y.; et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 2022, 28, 1679–1692. [Google Scholar] [CrossRef]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Kral, B.G.; Mathias, R.A.; Suktitipat, B.; Ruczinski, I.; Vaidya, D.; Yanek, L.R.; Quyyumi, A.A.; Patel, R.S.; Zafari, A.M.; Vaccarino, V.; et al. A common variant in the CDKN2B gene on chromosome 9p21 protects against coronary artery disease in Americans of African ancestry. J. Hum. Genet. 2011, 56, 224–229. [Google Scholar] [CrossRef]
- Schunkert, H.; Konig, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.; Barbalic, M.; Gieger, C.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef]
- Peden, J.F.; Hopewell, J.C.; Saleheen, D.; Chambers, J.C.; Hager, J.; Soranzo, N.; Collins, R.; Danesh, J.; Elliott, P.; Farrall, M.; et al. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011, 43, 339–344. [Google Scholar] [CrossRef]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Howson, J.M.M.; Zhao, W.; Barnes, D.R.; Ho, W.K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L.; et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Tan, V.Y.; Timpson, N.J. The UK Biobank: A Shining Example of Genome-Wide Association Study Science with the Power to Detect the Murky Complications of Real-World Epidemiology. Annu. Rev. Genom. Hum. Genet. 2022, 23, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Concato, J.; Brophy, M.; Fiore, L.; Pyarajan, S.; Breeling, J.; Whitbourne, S.; Deen, J.; Shannon, C.; Humphries, D.; et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 2016, 70, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Erdmann, J.; Stirrups, K.E.; Stitziel, N.O.; Masca, N.G.; Jansen, H.; Kanoni, S.; Nelson, C.P.; Ferrario, P.G.; Konig, I.R.; et al. Systematic Evaluation of Pleiotropy Identifies 6 Further Loci Associated with Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 823–836. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cheng, P.; Travisano, S.; Weldy, C.; Monteiro, J.P.; Kundu, R.; Nguyen, T.; Sharma, D.; Shi, H.; Lin, Y.; et al. Molecular mechanisms of coronary artery disease risk at the PDGFD locus. Nat. Commun. 2023, 14, 847. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Wunnemann, F.; Fotsing Tadjo, T.; Beaudoin, M.; Lalonde, S.; Lo, K.S.; Kleinstiver, B.P.; Lettre, G. Multimodal CRISPR perturbations of GWAS loci associated with coronary artery disease in vascular endothelial cells. PLoS Genet. 2023, 19, e1010680. [Google Scholar] [CrossRef]
- Xu, S.; Xu, Y.; Liu, P.; Zhang, S.; Liu, H.; Slavin, S.; Kumar, S.; Koroleva, M.; Luo, J.; Wu, X.; et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur. Heart J. 2019, 40, 2398–2408. [Google Scholar] [CrossRef]
- Lalonde, S.; Codina-Fauteux, V.A.; de Bellefon, S.M.; Leblanc, F.; Beaudoin, M.; Simon, M.M.; Dali, R.; Kwan, T.; Lo, K.S.; Pastinen, T.; et al. Integrative analysis of vascular endothelial cell genomic features identifies AIDA as a coronary artery disease candidate gene. Genome Biol. 2019, 20, 133. [Google Scholar] [CrossRef]
- Krause, M.D.; Huang, R.T.; Wu, D.; Shentu, T.P.; Harrison, D.L.; Whalen, M.B.; Stolze, L.K.; Di Rienzo, A.; Moskowitz, I.P.; Civelek, M.; et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc. Natl. Acad. Sci. USA 2018, 115, E11349–E11358. [Google Scholar] [CrossRef]
- Erdmann, J.; Willenborg, C.; Nahrstaedt, J.; Preuss, M.; König, I.R.; Baumert, J.; Linsel-Nitschke, P.; Gieger, C.; Tennstedt, S.; Belcredi, P.; et al. Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur. Heart J. 2010, 32, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Huang, R.T.; Kuo, C.H.; Kumar, S.; Kim, C.W.; Lin, Y.C.; Chen, Y.J.; Birukova, A.; Birukov, K.G.; Dulin, N.O.; et al. Mechanosensitive PPAP2B Regulates Endothelial Responses to Atherorelevant Hemodynamic Forces. Circ. Res. 2015, 117, e41–e53. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Civelek, M.; Fang, Y.; Fleming, I. The atherosusceptible endothelium: Endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 2013, 99, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Tchantchou, F.G.M.; Falcone, D.; Shea, T.B. S-adenosylmethionine mediates glutathione efficacy by increasing glutathione S-transferase activity: Implications for S-adenosyl methionine as a neuroprotec- tive dietary supplement. J. Alzheimers Dis. 2008, 14, 323–328. [Google Scholar] [CrossRef]
- Harris, T.J.; Tepass, U. Adherens junctions: From molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef]
- Kaufmann, U.; Zuppinger, C.; Waibler, Z.; Rudiger, M.; Urbich, C.; Martin, B.; Jockusch, B.M.; Eppenberger, H.; Starzinski-Powitz, A. The armadillo repeat region targets ARVCF to cadherin-based cellular junctions. J. Cell Sci. 2000, 113, 4121–4135. [Google Scholar] [CrossRef]
- Wei, P.; Milbauer, L.C.; Enenstein, J.; Nguyen, J.; Pan, W.; Hebbel, R.P. Differential endothelial cell gene expression by African Americans versus Caucasian Americans: A possible contribution to health disparity in vascular disease and cancer. BMC Med. 2011, 9, 2. [Google Scholar] [CrossRef]
- Hanley, P.J.; Xu, Y.; Kronlage, M.; Grobe, K.; Schon, P.; Song, J.; Sorokin, L.; Schwab, A.; Bahler, M. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc. Natl. Acad. Sci. USA 2010, 107, 12145–12150. [Google Scholar] [CrossRef]
- Yang, X.; Yang, W.; McVey, D.G.; Zhao, G.; Hu, J.; Poston, R.N.; Ren, M.; Willeit, K.; Coassin, S.; Willeit, J.; et al. FURIN Expression in Vascular Endothelial Cells Is Modulated by a Coronary Artery Disease-Associated Genetic Variant and Influences Monocyte Transendothelial Migration. J. Am. Heart Assoc. 2020, 9, e014333. [Google Scholar] [CrossRef] [PubMed]
- Stolze, L.K.; Conklin, A.C.; Whalen, M.B.; Rodríguez, M.L.; Õunap, K.; Selvarajan, I.; Toropainen, A.; Örd, T.; Li, J.; Eshghi, A.; et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am. J. Hum. Genet. 2020, 106, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Karamanavi, E.; McVey, D.G.; van der Laan, S.W.; Stanczyk, P.J.; Morris, G.E.; Wang, Y.; Yang, W.; Chan, K.; Poston, R.N.; Luo, J.; et al. The FES Gene at the 15q26 Coronary-Artery-Disease Locus Inhibits Atherosclerosis. Circ. Res. 2022, 131, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Depuydt, M.A.C.; Prange, K.H.M.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.A.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Cheng, P.; Wirka, R.C.; Shoa Clarke, L.; Zhao, Q.; Kundu, R.; Nguyen, T.; Nair, S.; Sharma, D.; Kim, H.J.; Shi, H.; et al. ZEB2 Shapes the Epigenetic Landscape of Atherosclerosis. Circulation 2022, 145, 469–485. [Google Scholar] [CrossRef]
- Nurnberg, S.T.; Guerraty, M.A.; Wirka, R.C.; Rao, H.S.; Pjanic, M.; Norton, S.; Serrano, F.; Perisic, L.; Elwyn, S.; Pluta, J.; et al. Genomic profiling of human vascular cells identifies TWIST1 as a causal gene for common vascular diseases. PLoS Genet. 2020, 16, e1008538. [Google Scholar] [CrossRef]
- Aherrahrou, R.; Guo, L.; Nagraj, V.P.; Aguhob, A.; Hinkle, J.; Chen, L.; Yuhl Soh, J.; Lue, D.; Alencar, G.F.; Boltjes, A.; et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ. Res. 2020, 127, 1552–1565. [Google Scholar] [CrossRef]
- Chignon, A.; Mathieu, S.; Rufiange, A.; Argaud, D.; Voisine, P.; Bosse, Y.; Arsenault, B.J.; Theriault, S.; Mathieu, P. Enhancer promoter interactome and Mendelian randomization identify network of druggable vascular genes in coronary artery disease. Hum. Genom. 2022, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Harismendy, O.; Notani, D.; Song, X.; Rahim, N.G.; Tanasa, B.; Heintzman, N.; Ren, B.; Fu, X.D.; Topol, E.J.; Rosenfeld, M.G.; et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 2011, 470, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Motterle, A.; Pu, X.; Wood, H.; Xiao, Q.; Gor, S.; Ng, F.L.; Chan, K.; Cross, F.; Shohreh, B.; Poston, R.N.; et al. Functional analyses of coronary artery disease associated variation on chromosome 9p21 in vascular smooth muscle cells. Hum. Mol. Genet. 2012, 21, 4021–4029. [Google Scholar] [CrossRef]
- Lo Sardo, V.; Chubukov, P.; Ferguson, W.; Kumar, A.; Teng, E.L.; Duran, M.; Zhang, L.; Cost, G.; Engler, A.J.; Urnov, F.; et al. Unveiling the Role of the Most Impactful Cardiovascular Risk Locus through Haplotype Editing. Cell 2018, 175, 1796–1810.e20. [Google Scholar] [CrossRef] [PubMed]
- Trillhaase, A.; Schmidt, B.; Märtens, M.; Haferkamp, U.; Erdmann, J.; Aherrahrou, Z. The CAD risk locus 9p21 increases the risk of vascular calcification in an iPSC-derived VSMC model. Stem Cell Res. Ther. 2021, 12, 166. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Lighthouse, J.K.; Greif, D.M. Development and pathologies of the arterial wall. Cell. Mol. Life Sci. 2014, 71, 1977–1999. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, K.K.; Rohovsky, S.A.; D’Amore, P.A. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 1998, 141, 805–814. [Google Scholar] [CrossRef]
- Low, E.L.; Baker, A.H.; Bradshaw, A.C. TGFβ, smooth muscle cells and coronary artery disease: A review. Cell Signal 2019, 53, 90–101. [Google Scholar] [CrossRef]
- Tsai, S.; Hollenbeck, S.T.; Ryer, E.J.; Edlin, R.; Yamanouchi, D.; Kundi, R.; Wang, C.; Liu, B.; Kent, K.C. TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H540–H549. [Google Scholar] [CrossRef]
- Iyer, D.; Zhao, Q.; Wirka, R.; Naravane, A.; Nguyen, T.; Liu, B.; Nagao, M.; Cheng, P.; Miller, C.L.; Kim, J.B.; et al. Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 2018, 14, e1007681. [Google Scholar] [CrossRef]
- Fardi, M.; Alivand, M.; Baradaran, B.; Hagh, M.F.; Solali, S. The crucial role of ZEB2: From development to epithelial-to-mesenchymal transition and cancer complexity. J. Cell Physiol. 2019, 234, 14783–14799. [Google Scholar] [CrossRef]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef]
- Mahmoud, M.M.; Kim, H.R.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Serbanovic-Canic, J.; Feng, S.; Bowden, N.P.; Maguire, R.; et al. TWIST1 Integrates Endothelial Responses to Flow in Vascular Dysfunction and Atherosclerosis. Circ. Res. 2016, 119, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yamashiro, K.; Ichikawa, Y.; Erlmann, P.; Kontani, K.; Malhotra, V.; Katada, T. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol. Biol. Cell 2011, 22, 2301–2308. [Google Scholar] [CrossRef]
- Nagao, M.; Lyu, Q.; Zhao, Q.; Wirka, R.C.; Bagga, J.; Nguyen, T.; Cheng, P.; Kim, J.B.; Pjanic, M.; Miano, J.M.; et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ. Res. 2020, 126, 517–529. [Google Scholar] [CrossRef]
- Soubeyrand, S.; Nikpay, M.; Turner, A.; Dang, A.T.; Herfkens, M.; Lau, P.; McPherson, R. Regulation of MFGE8 by the intergenic coronary artery disease locus on 15q26.1. Atherosclerosis 2019, 284, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Camare, C.; Pucelle, M.; Negre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noe, B. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.W.; Wong, D.; Dreisbach, C.N.; Miller, C.L. GWAS Reveal Targets in Vessel Wall Pathways to Treat Coronary Artery Disease. Front. Cardiovasc. Med. 2018, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Chauhan, S.K.; Kay, E.; Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7-dependent pathway. Blood 2011, 117, 5762–5771. [Google Scholar] [CrossRef]
- Jain, M.; Bhat, G.P.; Vijayraghavan, K.; Inamdar, M.S. Rudhira/BCAS3 is a cytoskeletal protein that controls Cdc42 activation and directional cell migration during angiogenesis. Exp. Cell Res. 2012, 318, 753–767. [Google Scholar] [CrossRef]
- Shetty, R.; Joshi, D.; Jain, M.; Vasudevan, M.; Paul, J.C.; Bhat, G.; Banerjee, P.; Abe, T.; Kiyonari, H.; VijayRaghavan, K.; et al. Rudhira/BCAS3 is essential for mouse development and cardiovascular patterning. Sci. Rep. 2018, 8, 5632. [Google Scholar] [CrossRef]
- Sapa-Wojciechowska, A.; Rak-Pasikowska, A.; Pormanczuk, K.; Czapla, B.; Bil-Lula, I. Extracellular Matrix Remodeling Factors as Markers of Carotid Artery Atherosclerosis. Cardiol. Res. Pract. 2020, 2020, 9036157. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Fisher, E.A. The double-edged sword of fibronectin in atherosclerosis. EMBO Mol. Med. 2012, 4, 561–563. [Google Scholar] [CrossRef]
- Feaver, R.E.; Gelfand, B.D.; Wang, C.; Schwartz, M.A.; Blackman, B.R. Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ. Res. 2010, 106, 1703–1711. [Google Scholar] [CrossRef]
- Rohwedder, I.; Montanez, E.; Beckmann, K.; Bengtsson, E.; Duner, P.; Nilsson, J.; Soehnlein, O.; Fassler, R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol. Med. 2012, 4, 564–576. [Google Scholar] [CrossRef]
- Soubeyrand, S.; Lau, P.; Nikpay, M.; Dang, A.T.; McPherson, R. Common Polymorphism That Protects From Cardiovascular Disease Increases Fibronectin Processing and Secretion. Circ. Genom. Precis. Med. 2022, 15, e003428. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Tesmenitsky, Y.; Croce, K.; Travers, R.; Shvartz, E.; Koskinas, K.C.; Sukhova, G.K.; Aikawa, E.; Aikawa, M.; Libby, P. Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2464–2472. [Google Scholar] [CrossRef]
- Pickrell, J.K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014, 94, 559–573. [Google Scholar] [CrossRef]
- Jarray, R.; Allain, B.; Borriello, L.; Biard, D.; Loukaci, A.; Larghero, J.; Hadj-Slimane, R.; Garbay, C.; Lepelletier, Y.; Raynaud, F. Depletion of the novel protein PHACTR-1 from human endothelial cells abolishes tube formation and induces cell death receptor apoptosis. Biochimie 2011, 93, 1668–1675. [Google Scholar] [CrossRef]
- Allain, B.; Jarray, R.; Borriello, L.; Leforban, B.; Dufour, S.; Liu, W.Q.; Pamonsinlapatham, P.; Bianco, S.; Larghero, J.; Hadj-Slimane, R.; et al. Neuropilin-1 regulates a new VEGF-induced gene, Phactr-1, which controls tubulogenesis and modulates lamellipodial dynamics in human endothelial cells. Cell Signal 2012, 24, 214–223. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Koplev, S.; Seldin, M.; Sukhavasi, K.; Ermel, R.; Pang, S.; Zeng, L.; Bankier, S.; Di Narzo, A.; Cheng, H.; Meda, V.; et al. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 2022, 1, 85–100. [Google Scholar] [CrossRef]
- Liu, B.; Pjanic, M.; Wang, T.; Nguyen, T.; Gloudemans, M.; Rao, A.; Castano, V.G.; Nurnberg, S.; Rader, D.J.; Elwyn, S.; et al. Genetic Regulatory Mechanisms of Smooth Muscle Cells Map to Coronary Artery Disease Risk Loci. Am. J. Hum. Genet. 2018, 103, 377–388. [Google Scholar] [CrossRef]
- Nanda, V.; Wang, T.; Pjanic, M.; Liu, B.; Nguyen, T.; Matic, L.P.; Hedin, U.; Koplev, S.; Ma, L.; Franzén, O.; et al. Functional regulatory mechanism of smooth muscle cell-restricted LMOD1 coronary artery disease locus. PLoS Genet. 2018, 14, e1007755. [Google Scholar] [CrossRef]
- Miller, C.L.; Pjanic, M.; Wang, T.; Nguyen, T.; Cohain, A.; Lee, J.D.; Perisic, L.; Hedin, U.; Kundu, R.K.; Majmudar, D.; et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 2016, 7, 12092. [Google Scholar] [CrossRef]
- Aherrahrou, R.; Lue, D.; Perry, R.N.; Aberra, Y.T.; Khan, M.D.; Soh, J.Y.; Örd, T.; Singha, P.; Yang, Q.; Gilani, H.; et al. Genetic Regulation of SMC Gene Expression and Splicing Predict Causal CAD Genes. Circ. Res. 2023, 132, 323–338. [Google Scholar] [CrossRef] [PubMed]
- McAfee, J.C.; Bell, J.L.; Krupa, O.; Matoba, N.; Stein, J.L.; Won, H. Focus on your locus with a massively parallel reporter assay. J. Neurodev. Disord. 2022, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Griesemer, D.; Xue, J.R.; Reilly, S.K.; Ulirsch, J.C.; Kukreja, K.; Davis, J.R.; Kanai, M.; Yang, D.K.; Butts, J.C.; Guney, M.H.; et al. Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell 2021, 184, 5247–5260.e19. [Google Scholar] [CrossRef] [PubMed]
- Myint, L.; Wang, R.; Boukas, L.; Hansen, K.D.; Goff, L.A.; Avramopoulos, D. A screen of 1049 schizophrenia and 30 Alzheimer’s-associated variants for regulatory potential. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020, 183, 61–73. [Google Scholar] [CrossRef]
- Zhao, S.; Hong, C.K.Y.; Myers, C.A.; Granas, D.M.; White, M.A.; Corbo, J.C.; Cohen, B.A. A single-cell massively parallel reporter assay detects cell-type-specific gene regulation. Nat. Genet. 2023, 55, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryń, Ł.M.; Rath, M.; Stark, A. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Toropainen, A.; Stolze, L.K.; Örd, T.; Whalen, M.B.; Torrell, P.M.; Link, V.M.; Kaikkonen, M.U.; Romanoski, C.E. Functional noncoding SNPs in human endothelial cells fine-map vascular trait associations. Genome Res. 2022, 32, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.W.; Dai, X.Y.; Wang, W.T.; Yang, Z.X.; Zhao, J.J.; Zhang, J.P.; Wen, W.; Zhang, F.; Oberg, K.C.; Zhang, L.; et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021, 49, 969–985. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Tay, K.Y.; Wu, K.X.; Chioh, F.W.J.; Autio, M.I.; Pek, N.M.Q.; Narmada, B.C.; Tan, S.-H.; Low, A.F.-H.; Lian, M.M.; Chew, E.G.Y.; et al. Trans-interaction of risk loci 6p24.1 and 10q11.21 is associated with endothelial damage in coronary artery disease. Atherosclerosis 2022, 362, 11–22. [Google Scholar] [CrossRef]
- Rees, H.A.; Yeh, W.-H.; Liu, D.R. Development of hRad51–Cas9 nickase fusions that mediate HDR without double-stranded breaks. Nat. Commun. 2019, 10, 2212. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Qu, Y.; Cheng, J.K.W.; Hughes, N.W.; Zhang, Q.; Wang, M.; Cong, L. dCas9-based gene editing for cleavage-free genomic knock-in of long sequences. Nat. Cell Biol. 2022, 24, 268–278. [Google Scholar] [CrossRef]
- Jensen, T.I.; Mikkelsen, N.S.; Gao, Z.; Foßelteder, J.; Pabst, G.; Axelgaard, E.; Laustsen, A.; König, S.; Reinisch, A.; Bak, R.O. Targeted regulation of transcription in primary cells using CRISPRa and CRISPRi. Genome Res. 2021, 31, 2120–2130. [Google Scholar] [CrossRef]
- Yuan, Q.; Gao, X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 2022, 13, 2771. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef]
- Soubeyrand, S.; Nikpay, M.; Lau, P.; Turner, A.; Hoang, H.D.; Alain, T.; McPherson, R. CARMAL Is a Long Non-coding RNA Locus That Regulates MFGE8 Expression. Front. Genet. 2020, 11, 631. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.-R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; He, S.; Huang, S.; Li, C.; Chen, Y.; Liu, Z.; Huang, X.; Wang, X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020, 6, 27. [Google Scholar] [CrossRef]
- Sakata, R.C.; Ishiguro, S.; Mori, H.; Tanaka, M.; Tatsuno, K.; Ueda, H.; Yamamoto, S.; Seki, M.; Masuyama, N.; Nishida, K.; et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020, 38, 865–869. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Solomon, C.U.; McVey, D.G.; Andreadi, C.; Gong, P.; Turner, L.; Stanczyk, P.J.; Khemiri, S.; Chamberlain, J.C.; Yang, W.; Webb, T.R.; et al. Effects of Coronary Artery Disease-Associated Variants on Vascular Smooth Muscle Cells. Circulation 2022, 146, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, X.; Zuo, G.; Pu, J.; Wu, X.; Chen, S. The Role of Angiogenesis in Coronary Artery Disease: A Double-Edged Sword: Intraplaque Angiogenesis in Physiopathology and Therapeutic Angiogenesis for Treatment. Curr. Pharm. Des. 2018, 24, 451–464. [Google Scholar] [CrossRef]
- Ratner, M. Genentech discloses safety concerns over Avastin. Nat. Biotechnol. 2004, 22, 1198. [Google Scholar] [CrossRef]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Lead Variant | GTEx | STARNET | EC a | VSMC b | ||
|---|---|---|---|---|---|---|
| Artery | Gene | Artery | Gene | |||
| rs28435150 | Tibial | FHL3 | AOR | FHL3 | INPP5B | |
| rs61776719 | Tibial | FHL3 | MAM | FHL3 | FHL3 | |
| rs56170783 | PPAP2B | |||||
| rs11585169 | MAM | ENSA | ||||
| rs11204693 | Tibial | ARNT | MAM | ARNT | GOLPH3L | |
| rs12568757 | Tibial | ARNT | MAM | ARNT | CTSK | |
| rs10888395 | Tibial | CTSS | MAM | ARNT | CTSS | |
| rs1196456 | Tibial | SNX27 | TDRKH-AS1 | |||
| rs11810571 | Aorta | TDRKH-AS1 | AOR | ARNT | GOLPH3L | |
| rs12741323 | Tibial | NME7 | AOR | ATP1B1 | NME7 | |
| rs61806987 | Aorta | NME7 | ||||
| rs1057239 | KIAA0040 | |||||
| rs6700559 | Tibial | RP11-92G12.3 | AOR | DDX59 | DDX59-AS1 | |
| rs17163363 | Tibial | RP11-378J18.8 | ||||
| rs16986953 | Tibial | OSR1 | ||||
| rs6736093 | Tibial | RP11-399B17.1 | AOR | TMEM87B | TMEM87B | |
| rs148812085 | CARF | MAM | AC023271.1 | |||
| rs6804986 | Tibial | ZNF589 | AOR | ZNF589 | NME6 | |
| rs34759087 | MAM | SHISA5 | ||||
| rs6800032 | Tibial | PCCB | AOR | SLC35G2 | NCK1-DT | |
| rs185244 | Aorta | MRAS | AOR | ESYT3 | ||
| rs357494 | Aorta | ARHGEF26 | MAM | ARHGEF26 | ||
| rs4266144 | Coronary | LINC00881 | AOR | TIPARP | ||
| rs781663 | AOR | REST | REST | |||
| rs2127821 | Tibial | RP11-33B1.1 | AOR | AC093752.1 | ||
| rs13124853 | Tibial | ZNF827 | MAM | ZNF827 | ||
| rs6841581 | AOR | EDNRA | ||||
| rs374218 | Aorta | SNHG18 | AOR | SEMA5A | SNHG18 | |
| rs17263917 | Aorta | SEMA5A | AOR | CTD-2201E9.1 | ||
| rs2910686 | Tibial | ERAP2 | AOR | ERAP2 | ERAP2 | |
| rs112949822 | AOR | FER | ||||
| rs9349379 | Tibial | PHACTR1 | MAM | GFOD1 | ||
| rs1034246 | Aorta | PTK7 | ||||
| rs9443626 | Tibial | IRAK1BP1 | AOR | IRAK1BP1 | IRAK1BP1 | |
| rs35510806 | CENPW | |||||
| rs2492304 | Tibial | SLC2A12 | ||||
| rs10951983 | Tibial | DAGLB | DAGLB | |||
| rs1019307 | Tibial | TMEM106B | TMEM106B | |||
| rs2107595 | Aorta | TWIST1 | MAM | AC003986.6 | ||
| rs17142613 | MACC1 | |||||
| rs2215614 | AOR | TBX20 | ||||
| rs56408342 | Aorta | BMP1 | AOR | BMP1 | ||
| rs17566555 | Tibial | CAMK1D | ||||
| rs9337951 | MAM | KIAA1462 | ||||
| rs55753709 | Tibial | PLCE1-AS1 | ||||
| rs884811 | AOR | LOXL4 | ||||
| rs6598075 | Tibial | RP11-326C3.16 | AOR | RIC8A | RIC8A | |
| rs360153 | MAM | SWAP70 | ||||
| rs633185 | Aorta | ARHGAP42 | MAM | ARHGAP42 | ||
| rs4754694 | Tibial | TMEM133 | MAM | ARHGAP42 | ARHGAP42 | |
| rs2839812 | Aorta | PDGFD | AOR | PDGFD | PDGFD | |
| rs1177562 | Tibial | HMBS | MAM | VPS11 | AP003392.4 | |
| rs17813323 | Tibial | YEATS4 | YEATS4 | |||
| rs2681472 | Tibial | ATP2B1 | MAM | ATP2B1 | ||
| rs11107903 | Aorta | FGD6 | AOR | FGD6 | FGD6 | |
| rs7133378 | Tibial | DNAH10OS | CCDC92 | |||
| rs7991314 | Aorta | N4BP2L2 | MAM | ATP8A2P2 | ||
| rs712486 | Tibial | HAUS4 | AOR | HAUS4 | ||
| rs10131894 | Tibial | EIF2B2 | MAM | EIF2B2 | EIF2B2 | MLH3 |
| rs1043674 | Aorta | EIF2B2 | MAM | EIF2B2 | NEK9 | |
| rs4903284 | Aorta | EIF2B2 | MAM | EIF2B2 | EIF2B2 | |
| rs36033161 | Aorta | HHIPL1 | ||||
| rs7403103 | Tibial | TRIP4 | AOR | TRIP4 | TRIP4 | |
| rs56062135 | SMAD3 | |||||
| rs62011052 | Aorta | ADAMTS7 | AOR | ADAMTS7 | ADAMTS7 | |
| rs7173743 | Aorta | ADAMTS7 | MAM | ADAMTS7 | ||
| rs1807214 | Aorta | HAPLN3 | AOR | ABHD2 | ||
| rs8032315 | Aorta | FES | FES | |||
| rs7183988 | Aorta | FURIN | FES | FES | ||
| rs1894400 | Aorta | FES | FES | |||
| rs1800775 | AOR | AMFR | CETP | |||
| rs7195958 | Tibial | DHODH | MAM | DHODH | DHODH | |
| rs1050362 | Tibial | DHODH | AOR | DHX38 | DHX38 | |
| rs8046696 | Tibial | BCAR1 | AOR | BCAR1 | CFDP1 | |
| rs7500448 | Aorta | CDH13 | AOR | CDH13 | ||
| rs4790881 | MAM | SMG6 | ||||
| rs12936927 | Aorta | AC122129.1 | TOM1L2 | |||
| rs7207292 | Tibial | EFCAB5 | AOR | EFCAB5 | EFCAB5 | |
| rs2074164 | Tibial | DHX58 | MAM | DHX58 | DHX58 | |
| rs4792923 | Tibial | NAGLU | MAM | NAGLU | NAGLU | |
| rs5820757 | Tibial | ZNF652 | ||||
| rs4794006 | Tibial | SUMO2P17 | MAM | UBE2Z | ATP5MC1 | |
| rs11079536 | Tibial | TEX2 | PECAM1 | |||
| rs2909217 | Tibial | PRKAR1A | ||||
| rs11663411 | Tibial | LMAN1 | LMAN1 | |||
| rs35562870 | Tibial | MARCH2 | MAM | MARCH2 | MARCH2 | |
| rs7246865 | Tibial | MYO9B | ||||
| rs10410487 | MAP1S | |||||
| rs2972445 | Aorta | ZNF571-AS1 | MAM | ZFP30 | ZFP30 | |
| rs10409487 | Tibial | CTD-3220F14.3 | MAM | ZFP30 | ZFP30 | |
| rs11466359 | Tibial | AXL | AXL | |||
| rs1800469 | Tibial | B9D2 | ||||
| rs2241709 | Aorta | EXOSC5 | MAM | DMAC2 | BCKDHA | |
| rs8108474 | AOR | DMPK | ||||
| rs73354869 | Tibial | LINC00189 | MAP3K7CL | |||
| rs28451064 | AOR | AP000318.2 | ||||
| rs35219138 | Tibial | PDXK | RRP1B | |||
| rs71313931 | Aorta | ARVCF | AOR | ARVCF | ARVCF | |
| rs468224 | Tibial | THOC5 | AOR | THOC5 | THOC5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quaye, L.N.K.; Dalzell, C.E.; Deloukas, P.; Smith, A.J.P. The Genetics of Coronary Artery Disease: A Vascular Perspective. Cells 2023, 12, 2232. https://doi.org/10.3390/cells12182232
Quaye LNK, Dalzell CE, Deloukas P, Smith AJP. The Genetics of Coronary Artery Disease: A Vascular Perspective. Cells. 2023; 12(18):2232. https://doi.org/10.3390/cells12182232
Chicago/Turabian StyleQuaye, Leon N. K., Catherine E. Dalzell, Panos Deloukas, and Andrew J. P. Smith. 2023. "The Genetics of Coronary Artery Disease: A Vascular Perspective" Cells 12, no. 18: 2232. https://doi.org/10.3390/cells12182232