
Obatoclax Rescues FUS-ALS Phenotypes in iPSC-Derived Neurons by Inducing Autophagy

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. BH3 Mimetics
2.2. iPSC Culture and Assays
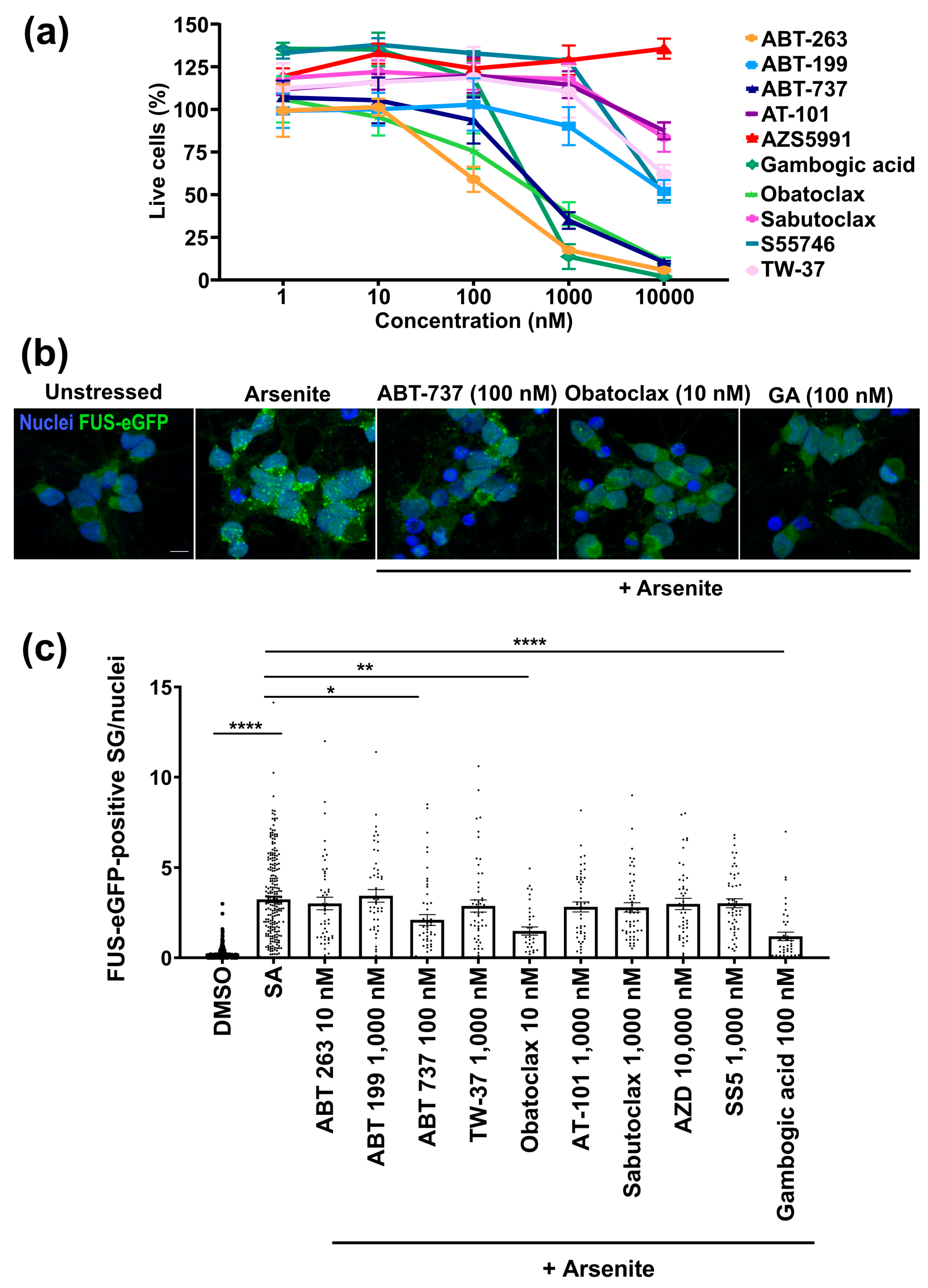
- To assay cell survival, neurons were seeded at 4 × 104 cells per well in a 96-well plate. After 20 days, neurons were treated with the compound at different concentrations (1, 10, 100, 1000, and 10,000 nM) for 24 h. After the treatment, the neurons were incubated with calcein-AM red (1 µM, Cayman, Ann Arbor, MI, USA) for 30 min. Then, wash two times with PBS (pH 7.5). The plate was read using a 560 nm excitation filter and a 590 nm emission filter on a Biotek Synergy™ NEO microplate reader (Agilent, Santa Clara, CA, USA). The fluorescence intensity is proportional to the number of viable cells.
- To assay stress granules, neurons seeded at 4 × 104 cells per well in a 96-well plate were treated with the compound at 10 nM for 24 h. After the treatment, the formation of stress granules was induced with the addition of sodium arsenite (500 mM, Sigma Sigma-Aldrich, St. Louis, MO, USA) for one hour.
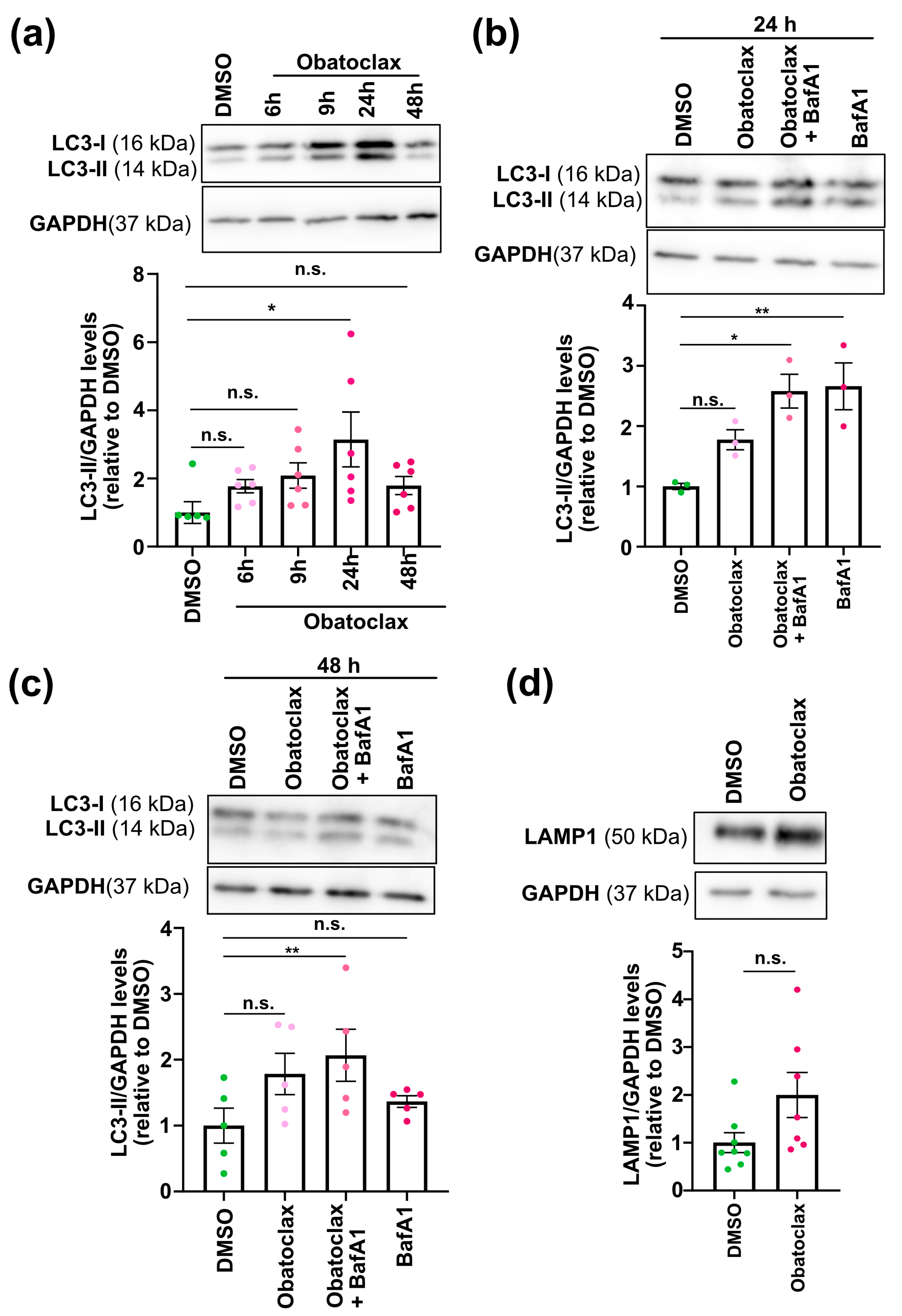
- To monitor autophagic flux, neurons seeded at 4 × 104 cells per well in a 96-well plate and 1.2 × 106 cells per well in a 12-well plate were treated with the compound at 10 nM for 6, 9, 24, and 48 h. Treatment with Bafilomycin A (10 nM, Selleckchem, Cologne, Germany) for 24 h was used as a control.
2.3. Protein Extraction, Immunoblotting, and Capillary Electrophoresis
2.4. Proximity Ligation Assay
2.5. Immunocytochemistry and Image Acquisition
2.6. Proteomics
2.7. Statistical Analysis
3. Results
3.1. Obatoclax Is Well-Tolerated and Potently Reduces Aberrant SG Formation
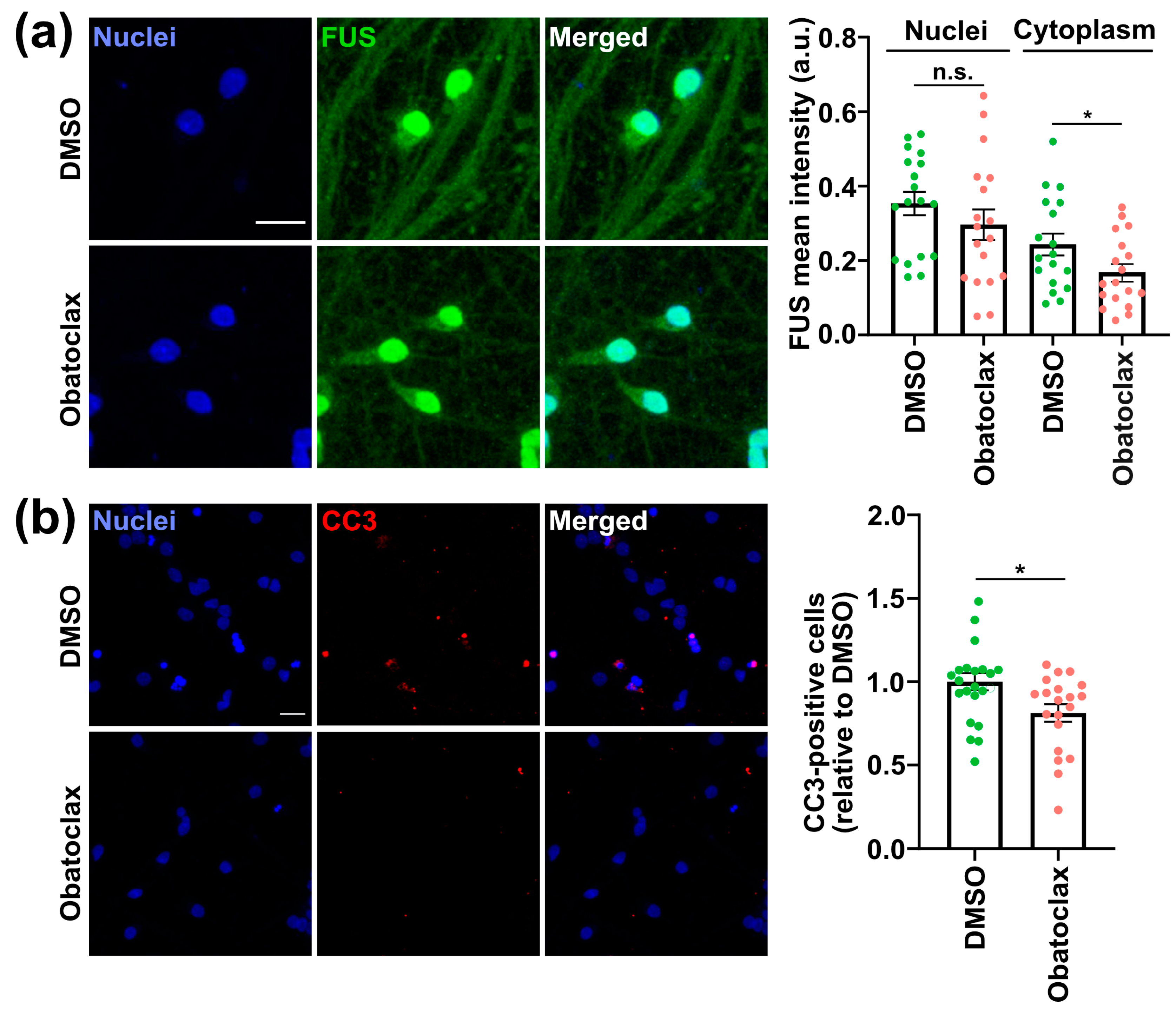
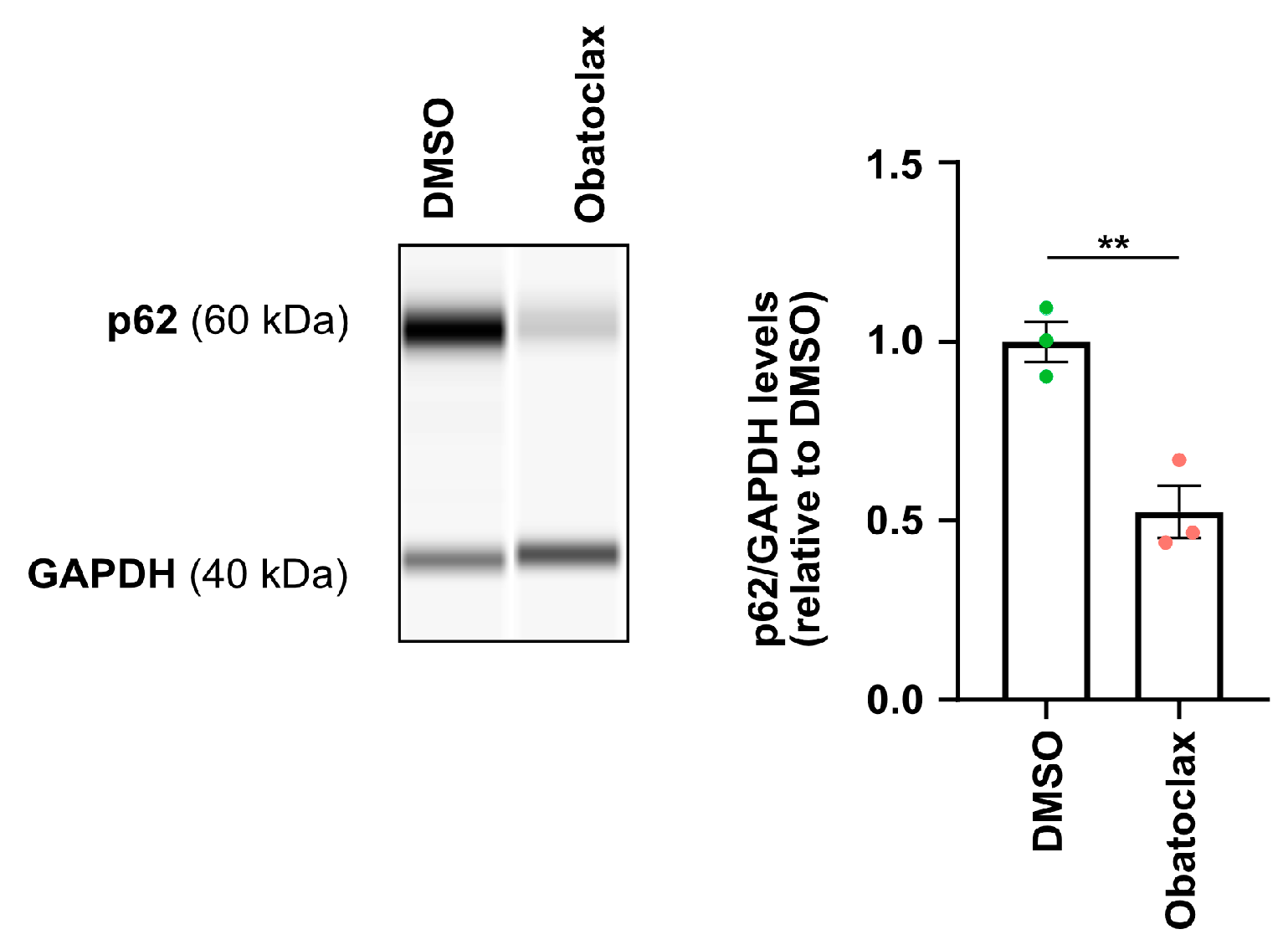
3.2. Obatoclax Reduces Cytoplasmic FUS Levels, Rescuing Aberrant Protein Homeostasis
3.3. Obatoclax Ameliorates the Degeneration of iPSC-Derived Neurons with Mutant FUS
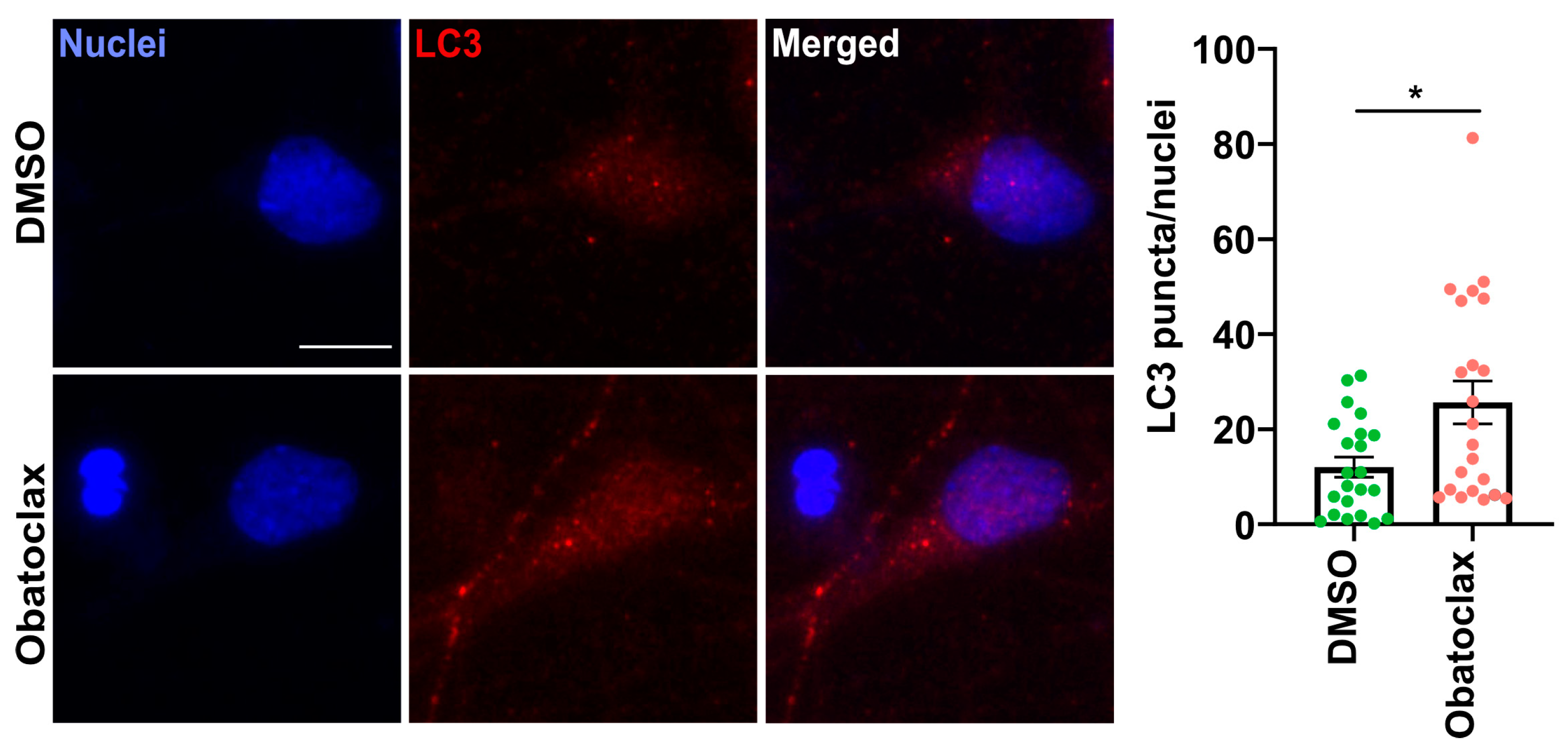
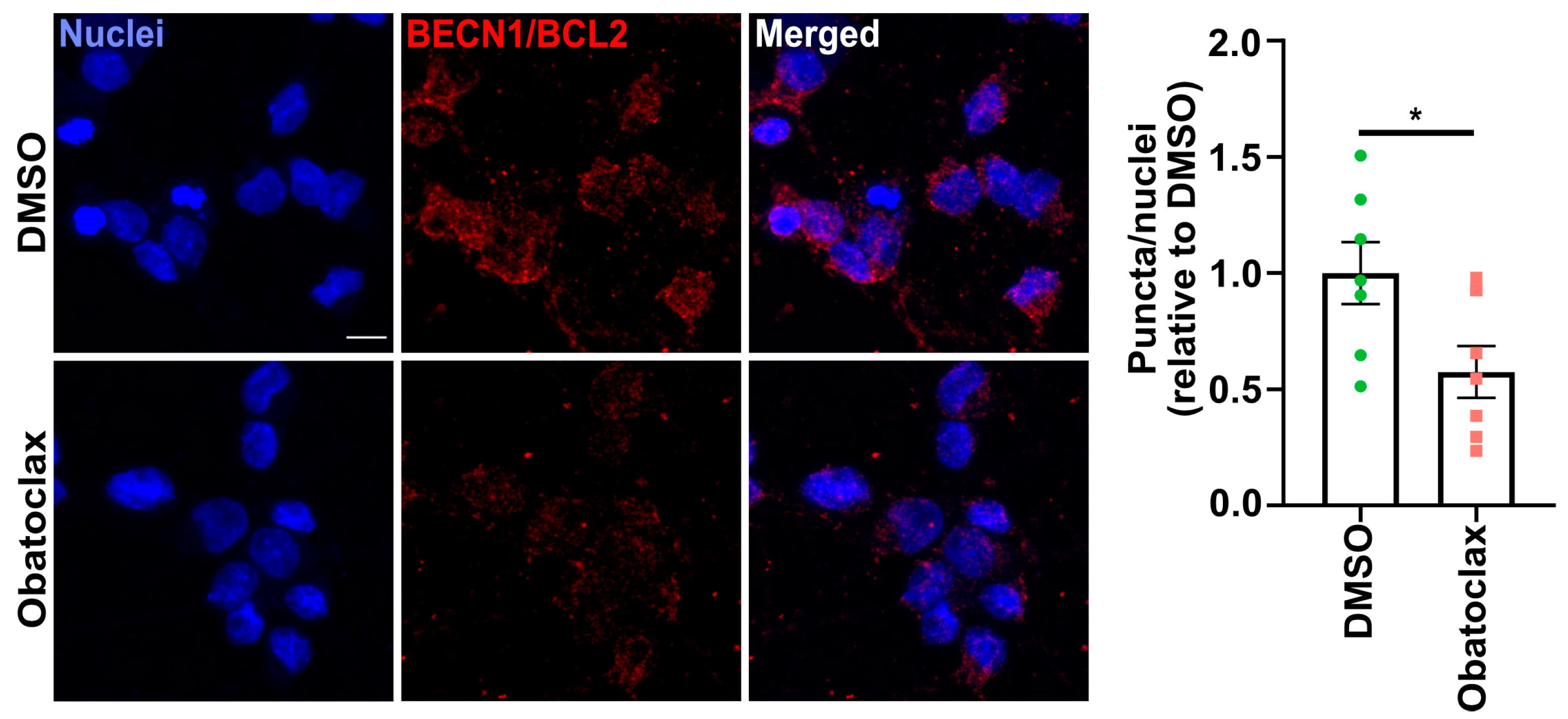
3.4. Obatoclax Induces Autophagy by Disrupting the BECN1-BCL2 Complex
3.5. Proteomics Suggests Obatoclax Contributes to Neuroprotection via Multiple Mechanisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef]
- Marrone, L.; Poser, I.; Casci, I.; Japtok, J.; Reinhardt, P.; Janosch, A.; Andree, C.; Lee, H.O.; Moebius, C.; Koerner, E.; et al. Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell Rep. 2018, 10, 375–389. [Google Scholar] [CrossRef] [PubMed]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Beclin 1: A BH3-only protein that fails to induce apoptosis. Oncogene 2009, 28, 2125–2127. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef]
- Bellmann, J.; Goswami, R.Y.; Girardo, S.; Rein, N.; Hosseinzadeh, Z.; Hicks, M.R.; Busskamp, V.; Pyle, A.D.; Werner, C.; Sterneckert, J. A customizable microfluidic platform for medium-throughput modeling of neuromuscular circuits. Biomaterials 2019, 225, 119537. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass Spectrometric Sequencing of Proteins from Silver-Stained Polyacrylamide Gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Vasilj, A.; Gentzel, M.; Ueberham, E.; Gebhardt, R.; Shevchenko, A. Tissue Proteomics by One-Dimensional Gel Electrophoresis Combined with Label-Free Protein Quantification. J. Proteome Res. 2012, 11, 3680–3689. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.A.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Dudman, J.; Qi, X. Stress Granule Dysregulation in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2020, 14, 598517. [Google Scholar] [CrossRef]
- Hwang, J.J.; Kuruvilla, J.; Mendelson, D.; Pishvaian, M.J.; Deeken, J.F.; Siu, L.L.; Berger, M.S.; Viallet, J.; Marshall, J.L. Phase I dose finding studies of obatoclax (GX15-070), a small molecule pan-BCL-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin. Cancer Res. 2010, 16, 4038–4045. [Google Scholar] [CrossRef]
- McCoy, F.; Hurwitz, J.; McTavish, N.; Paul, I.; Barnes, C.; O’Hagan, B.; Odrzywol, K.; Murray, J.; Longley, D.; McKerr, G.; et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis. 2010, 1, e108. [Google Scholar] [CrossRef]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef]
- Koehler, B.C.; Jassowicz, A.; Scherr, A.L.; Lorenz, S.; Radhakrishnan, P.; Kautz, N.; Elssner, C.; Weiss, J.; Jaeger, D.; Schneider, M.; et al. Pan-Bcl-2 inhibitor Obatoclax is a potent late stage autophagy inhibitor in colorectal cancer cells independent of canonical autophagy signaling. BMC Cancer 2015, 15, 919. [Google Scholar] [CrossRef] [PubMed]
- Daigle, J.G.; Lanson, N.A., Jr.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.J.; Sun, Z.K.; Shen, C.T.; Song, H.J.; Zhang, X.Y.; Qiu, Z.L.; Luo, Q.Y. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017, 7, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Soo, K.Y.; Sultana, J.; King, A.E.; Atkinson, R.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.; Farg, M.A.; Atkin, J.D. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. The autophagosome is overrated! Autophagy 2011, 7, 353–354. [Google Scholar] [CrossRef]
- Cheroni, C.; Marino, M.; Tortarolo, M.; Veglianese, P.; De Biasi, S.; Fontana, E.; Zuccarello, L.V.; Maynard, C.J.; Dantuma, N.P.; Bendotti, C. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 82–96. [Google Scholar] [CrossRef]
- Farrawell, N.E.; McAlary, L.; Lum, J.S.; Chisholm, C.G.; Warraich, S.T.; Blair, I.P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. Ubiquitin Homeostasis Is Disrupted in TDP-43 and FUS Cell Models of ALS. iScience 2020, 23, 101700. [Google Scholar] [CrossRef]
- Kabashi, E.; Agar, J.N.; Taylor, D.M.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef]
- Kabashi, E.; Agar, J.N.; Strong, M.J.; Durham, H.D. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 367–371. [Google Scholar] [CrossRef]
- Tashiro, Y.; Urushitani, M.; Inoue, H.; Koike, M.; Uchiyama, Y.; Komatsu, M.; Tanaka, K.; Yamazaki, M.; Abe, M.; Misawa, H.; et al. Motor Neuron-specific Disruption of Proteasomes, but Not Autophagy, Replicates Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2012, 287, 42984–42994. [Google Scholar] [CrossRef]
- Yoshihara, T.; Satake, H.; Nishie, T.; Okino, N.; Hatta, T.; Otani, H.; Naruse, C.; Suzuki, H.; Sugihara, K.; Kamimura, E.; et al. Lactosylceramide synthases encoded by B4galt5 and 6 genes are pivotal for neuronal generation and myelin formation in mice. PLoS Genet. 2018, 14, e1007545. [Google Scholar] [CrossRef] [PubMed]
- Nishie, T.; Hikimochi, Y.; Zama, K.; Fukusumi, Y.; Ito, M.; Yokoyama, H.; Naruse, C.; Ito, M.; Asano, M. Beta4-galactosyltransferase-5 is a lactosylceramide synthase essential for mouse extra-embryonic development. Glycobiology 2010, 20, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.; Lunghi, G.; Henriques, A.; Callizot, N.; Ciampa, M.G.; Mauri, L.; Prioni, S.; Carsana, E.V.; Loberto, N.; Aureli, M.; et al. GM1 Oligosaccharide Efficacy in Parkinson’s Disease: Protection against MPTP. Biomedicines 2023, 11, 1305. [Google Scholar] [CrossRef] [PubMed]
- Pestronk, A.; Adams, R.N.; Clawson, L.; Cornblath, D.; Kuncl, R.W.; Griffin, D.; Drachman, D.B. Serum antibodies to GM1 ganglioside in amyotrophic lateral sclerosis. Neurology 1988, 38, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.G.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside improves spatial learning and memory of aged rats. Behav. Brain Res. 1997, 85, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Naslavsky, N.; Caplan, S. A role for EHD4 in the regulation of early endosomal transport. Traffic 2008, 9, 995–1018. [Google Scholar] [CrossRef]
- Canagarajah, B.J.; Ren, X.; Bonifacino, J.S.; Hurley, J.H. The clathrin adaptor complexes as a paradigm for membrane-associated allostery. Protein Sci. 2013, 22, 517–529. [Google Scholar] [CrossRef]
- Rivero, S.; Rodríguez-Real, G.; Marín, I.; Huertas, P. MRGBP, a member of the NuA4 complex, inhibits DNA double-strand break repair. FEBS Open Bio 2021, 11, 622–632. [Google Scholar] [CrossRef]
- Hammond, C.M.; Bao, H.; Hendriks, I.A.; Carraro, M.; García-Nieto, A.; Liu, Y.; Reverón-Gómez, N.; Spanos, C.; Chen, L.; Rappsilber, J.; et al. DNAJC9 integrates heat shock molecular chaperones into the histone chaperone network. Mol. Cell 2021, 81, 2533–2548.e9. [Google Scholar] [CrossRef]
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897. [Google Scholar] [CrossRef]
- Chen, H.J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The heat shock response plays an important role in TDP-43 clearance: Evidence for dysfunction in amyotrophic lateral sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef]
- Mourelatos, Z.; Adler, H.; Hirano, A.; Donnenfeld, H.; Gonatas, J.O.; Gonatas, N.K. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. USA 1990, 87, 4393–4395. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, B.M.; Hartmann, H.; Serdaroglu, A.; Schludi, M.H.; Hornburg, D.; Meissner, F.; Orozco, D.; Colombo, A.; Tahirovic, S.; Michaelsen, M.; et al. TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 2016, 35, 2350–2370. [Google Scholar] [CrossRef] [PubMed]
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 2018, 12, 86. [Google Scholar] [CrossRef]
- van Dis, V.; Kuijpers, M.; Haasdijk, E.D.; Teuling, E.; Oakes, S.A.; Hoogenraad, C.C.; Jaarsma, D. Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol. Commun. 2014, 2, 38. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Gelino, S.; Chang, J.T.; Kumsta, C.; She, X.; Davis, A.; Nguyen, C.; Panowski, S.; Hansen, M. Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLOS Genet. 2016, 12, e1006135. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Fernandez, E.; Flurkey, K.; Han, M.; Javors, M.A.; Li, X.; Nadon, N.L.; Nelson, J.F.; et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 2014, 13, 468–477. [Google Scholar] [CrossRef]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [PubMed]
- Shahheydari, H.; Ragagnin, A.; Walker, A.K.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-L.; Duan, W.-J.; Lu, D.-H.; Ma, X.-H.; Li, X.-X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.-Z.; Li, Y.-F.; et al. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Zhang, S.; Duan, W.; Wei, R.; Chen, H.; Cai, W.; Yang, L.; Wang, Q. Enhanced Autophagy Contributes to Protective Effects of GM1 Ganglioside Against Aβ1-42-Induced Neurotoxicity and Cognitive Deficits. Neurochem. Res. 2017, 42, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Su, J.C.; Liu, C.Y.; Huang, J.W.; Chen, K.C.; Chen, W.L.; Tai, W.T.; Shiau, C.W. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett. 2012, 321, 27–35. [Google Scholar] [CrossRef]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C., Jr.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef]
- Trudel, S.; Li, Z.H.; Rauw, J.; Tiedemann, R.E.; Wen, X.Y.; Stewart, A.K. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 2007, 109, 5430–5438. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Claxton, D.F.; Crump, M.; Faderl, S.; Kipps, T.; Keating, M.J.; Viallet, J.; Cheson, B.D. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood 2009, 113, 299–305. [Google Scholar] [CrossRef]
- Schimmer, A.D.; O’Brien, S.; Kantarjian, H.; Brandwein, J.; Cheson, B.D.; Minden, M.D.; Yee, K.; Ravandi, F.; Giles, F.; Schuh, A.; et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 8295–8301. [Google Scholar] [CrossRef]
- Schlosser, A.; Volkmer-Engert, R. Volatile polydimethylcyclosiloxanes in the ambient laboratory air identified as source of extreme background signals in nanoelectrospray mass spectrometry. J. Mass. Spectrom. 2003, 38, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Walters, H.E.; Troyanovskiy, K.E.; Graf, A.M.; Yun, M.H. Senescent cells enhance newt limb regeneration by promoting muscle dedifferentiation. Aging Cell 2023, 22, e13826. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo Bautista, C.M.; Eismann, K.; Gentzel, M.; Pelucchi, S.; Mertens, J.; Walters, H.E.; Yun, M.H.; Sterneckert, J. Obatoclax Rescues FUS-ALS Phenotypes in iPSC-Derived Neurons by Inducing Autophagy. Cells 2023, 12, 2247. https://doi.org/10.3390/cells12182247
Castillo Bautista CM, Eismann K, Gentzel M, Pelucchi S, Mertens J, Walters HE, Yun MH, Sterneckert J. Obatoclax Rescues FUS-ALS Phenotypes in iPSC-Derived Neurons by Inducing Autophagy. Cells. 2023; 12(18):2247. https://doi.org/10.3390/cells12182247
Chicago/Turabian StyleCastillo Bautista, Cristina Marisol, Kristin Eismann, Marc Gentzel, Silvia Pelucchi, Jerome Mertens, Hannah E. Walters, Maximina H. Yun, and Jared Sterneckert. 2023. "Obatoclax Rescues FUS-ALS Phenotypes in iPSC-Derived Neurons by Inducing Autophagy" Cells 12, no. 18: 2247. https://doi.org/10.3390/cells12182247
APA StyleCastillo Bautista, C. M., Eismann, K., Gentzel, M., Pelucchi, S., Mertens, J., Walters, H. E., Yun, M. H., & Sterneckert, J. (2023). Obatoclax Rescues FUS-ALS Phenotypes in iPSC-Derived Neurons by Inducing Autophagy. Cells, 12(18), 2247. https://doi.org/10.3390/cells12182247