
Tumor Progression Reverses Cardiac Hypertrophy and Fibrosis in a Tetracycline-Regulated ATF3 Transgenic Mouse Model

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Cancer Implantation
2.4. Echocardiography
2.5. Fibrosis Staining
2.6. Cell Size Staining
2.7. RNA Extraction
2.8. qRT-PCR
2.9. Heart Single-Cell Suspension and Flow Cytometry
2.10. Macrophage Depletion
2.11. Statistics
3. Results
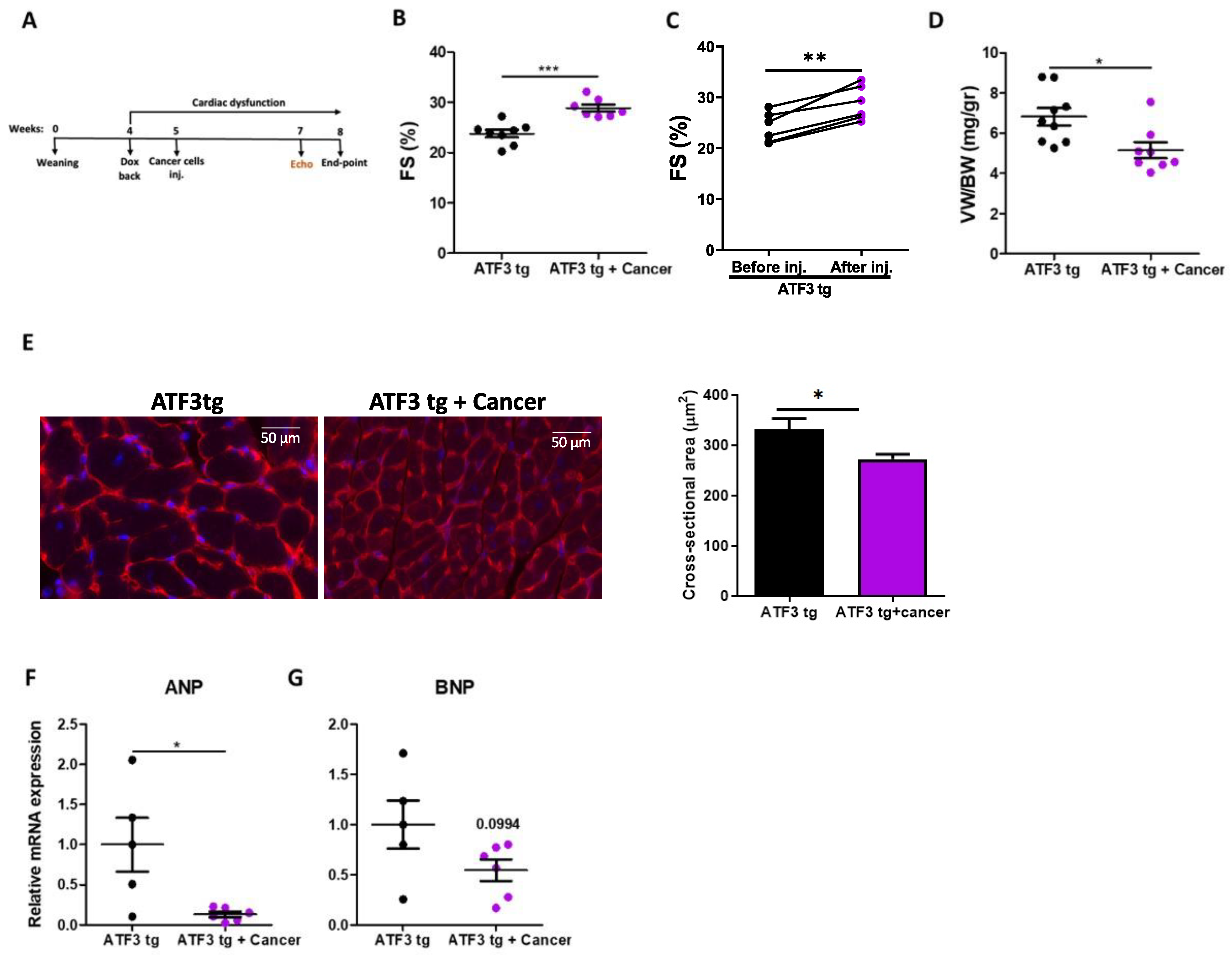
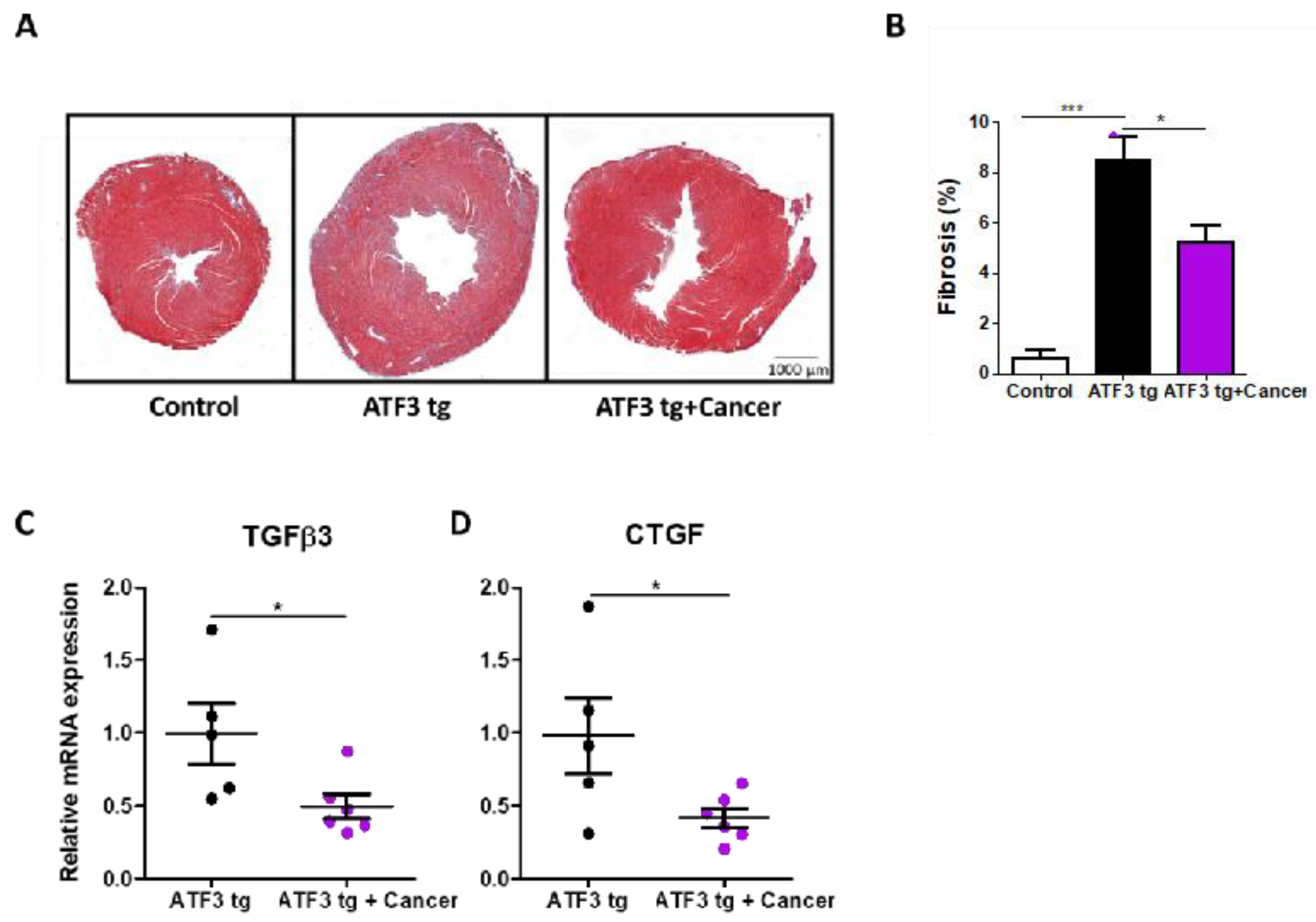
3.1. LLC Tumor Growth Ameliorates Cardiac Dysfunction in ATF3 Transgenic Mice
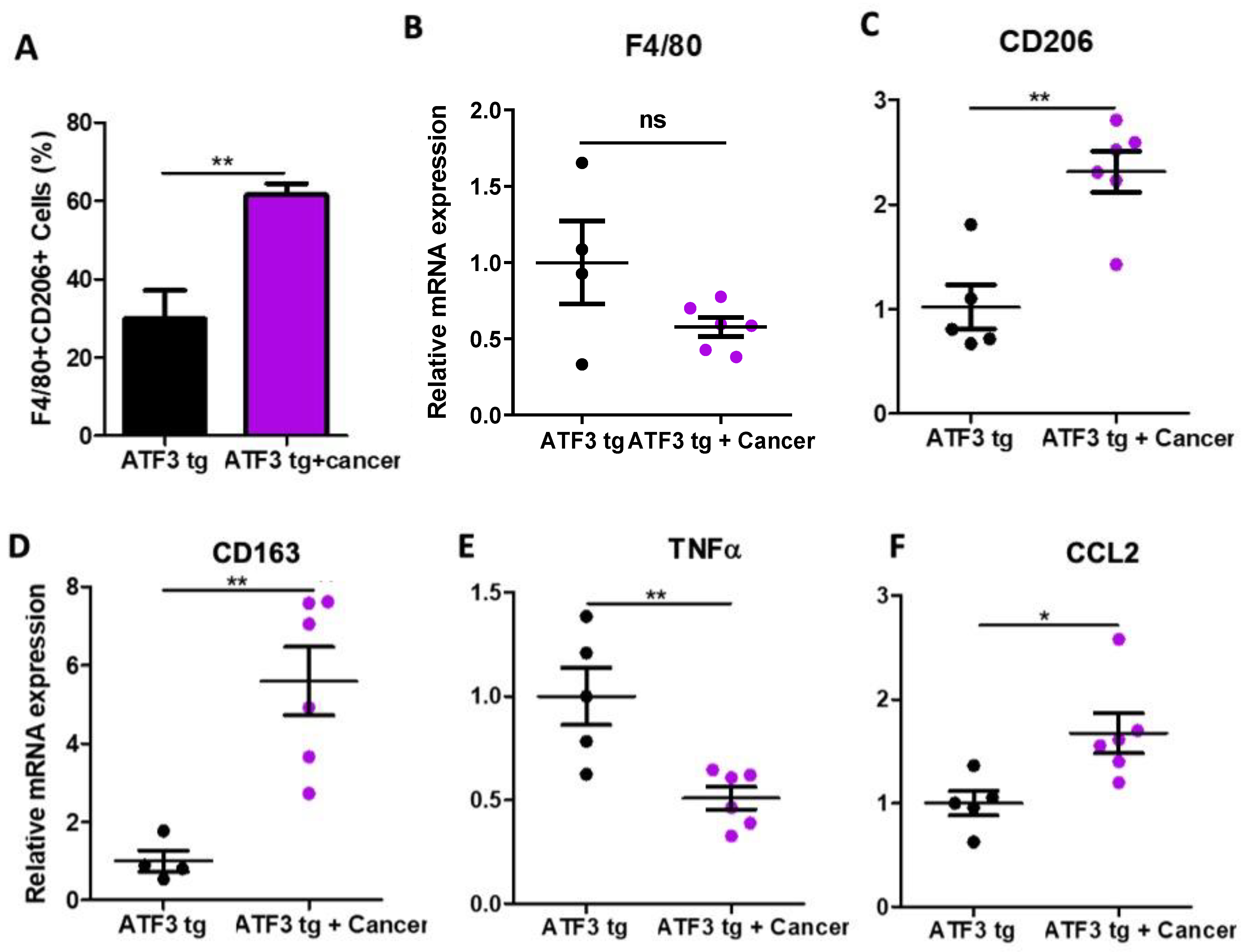
3.2. Tumor-Bearing Mice Exhibit M1 to M2 Macrophage Polarization in the Heart
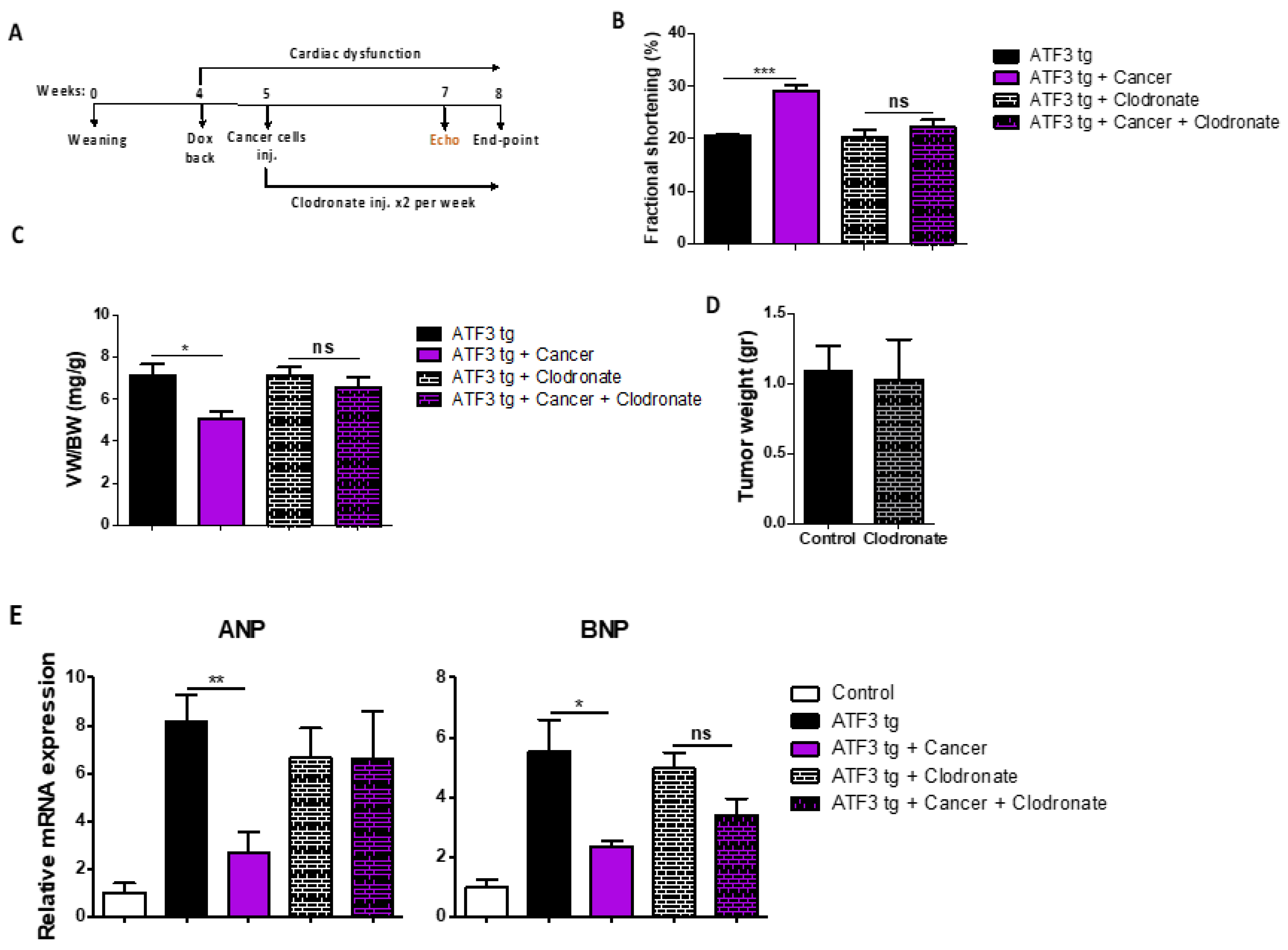
3.3. Macrophage Depletion Abrogates Tumor-Dependent Amelioration of Cardiac Dysfunction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Ausoni, S.; Azzarello, G. Development of Cancer in Patients With Heart Failure: How Systemic Inflammation Can Lay the Groundwork. Front. Cardiovasc. Med. 2020, 7, 598384. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.; Zhang, Q.; Moore, K.J. Crosstalk Between the Heart and Cancer: Beyond Drug Toxicity. Circulation 2020, 142, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell Cardiol. 2022, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van der Vegt, B.; et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef]
- Avraham, S.; Abu-Sharki, S.; Shofti, R.; Haas, T.; Korin, B.; Kalfon, R.; Friedman, T.; Shiran, A.; Saliba, W.; Shaked, Y.; et al. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation 2020, 142, 670–683. [Google Scholar] [CrossRef]
- Awwad, L.; Aronheim, A. Cardiac Dysfunction Promotes Cancer Progression via Multiple Secreted Factors. Cancer Res. 2022, 82, 1753–1761. [Google Scholar] [CrossRef]
- Awwad, L.; Goldenberg, T.; Langier-Goncalves, I.; Aronheim, A. Cardiac Remodeling in the Absence of Cardiac Contractile Dysfunction Is Sufficient to Promote Cancer Progression. Cells 2022, 11, 1108. [Google Scholar] [CrossRef]
- Hasin, T.; Gerber, Y.; Weston, S.A.; Jiang, R.; Killian, J.M.; Manemann, S.M.; Cerhan, J.R.; Roger, V.L. Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer. J. Am. Coll. Cardiol. 2016, 68, 265–271. [Google Scholar] [CrossRef]
- Saliba, W.; Bental, T.; Shapira, Y.; Schwartzenberg, S.; Sagie, A.; Vaturi, M.; Adawi, S.; Fuks, A.; Aronheim, A.; Shiran, A. Increased risk of non-hematological cancer in young patients with aortic stenosis: A retrospective cohort study. Cardiooncology 2021, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Awwad, L.; Shofti, R.; Haas, T.; Aronheim, A. Tumor growth ameliorates cardiac dysfunction. Cells 2023, 12, 1853. [Google Scholar] [CrossRef] [PubMed]
- Baumeier, C.; Escher, F.; Aleshcheva, G.; Pietsch, H.; Schultheiss, H.P. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res. Cardiol. 2021, 116, 1. [Google Scholar] [CrossRef] [PubMed]
- Kistner, A.; Gossen, M.; Zimmermann, F.; Jerecic, J.; Ullmer, C.; Lubbert, H.; Bujard, H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10933–10938. [Google Scholar] [CrossRef]
- Hai, T. (Ed.) The ATF Transcription Factors in Cellular Adaptive Responses; Higher Education Press: Beijing, China; Springer: New York, NY, USA, 2006; pp. 329–340. [Google Scholar]
- Hai, T.; Wolford, C.C.; Chang, Y.S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, D.F.; Bian, Z.Y.; Zong, J.; Deng, W.; Zhang, Y.; Guo, Y.Y.; Li, H.; Tang, Q.Z. Activating transcription factor 3 deficiency promotes cardiac hypertrophy, dysfunction, and fibrosis induced by pressure overload. PLoS ONE 2011, 6, e26744. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Zhang, C.; Li, P.; Wu, Y.; Wang, C.; Lau, W.B.; Ma, X.L.; Du, J. Cardiac Fibroblast-Specific Activating Transcription Factor 3 Protects Against Heart Failure by Suppressing MAP2K3-p38 Signaling. Circulation 2017, 135, 2041–2057. [Google Scholar] [CrossRef]
- Okamoto, Y.; Chaves, A.; Chen, J.; Kelley, R.; Jones, K.; Weed, H.G.; Gardner, K.L.; Gangi, L.; Yamaguchi, M.; Klomkleaw, W.; et al. Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am. J. Pathol. 2001, 159, 639–650. [Google Scholar] [CrossRef]
- Koren, L.; Elhanani, O.; Kehat, I.; Hai, T.; Aronheim, A. Adult Cardiac Expression of the Activating Transcription Factor 3, ATF3, Promotes Ventricular Hypertrophy. PLoS ONE 2013, 8, e68396. [Google Scholar] [CrossRef]
- Yu, Z.; Redfern, C.S.; Fishman, G.I. Conditional transgene expression in the heart. Circ. Res. 1996, 79, 691–697. [Google Scholar] [CrossRef]
- Avraham, S.; Korin, B.; Aviram, S.; Shechter, D.; Shaked, Y.; Aronheim, A. ATF3 and JDP2 deficiency in cancer associated fibroblasts promotes tumor growth via SDF-1 transcription. Oncogene 2019, 38, 3812–3823. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, L.; Epelman, S.; Clemente-Casares, X. Isolation and Identification of Extravascular Immune Cells of the Heart. J. Vis. Exp. 2018, 138, e58114. [Google Scholar] [CrossRef]
- Kalfon, R.; Friedman, T.; Eliachar, S.; Shofti, R.; Haas, T.; Koren, L.; Moskovitz, J.D.; Hai, T.; Aronheim, A. JDP2 and ATF3 deficiencies dampen maladaptive cardiac remodeling and preserve cardiac function. PLoS ONE 2019, 14, e0213081. [Google Scholar] [CrossRef]
- Jagannathan, R.; Patel, S.A.; Ali, M.K.; Narayan, K.M.V. Global Updates on Cardiovascular Disease Mortality Trends and Attribution of Traditional Risk Factors. Curr. Diabetes Rep. 2019, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Berliner, D.; Bauersachs, J. New drugs: Big changes in conservative heart failure therapy? Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2019, 55, i3–i10. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Achlaug, L.; Awwad, L.; Langier Goncalves, I.; Goldenberg, T.; Aronheim, A. Tumor growth ameliorates cardiac dysfunction and dampens fibrosis in a mouse model for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2023, 24, 12595. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awwad, L.; Aronheim, A. Tumor Progression Reverses Cardiac Hypertrophy and Fibrosis in a Tetracycline-Regulated ATF3 Transgenic Mouse Model. Cells 2023, 12, 2289. https://doi.org/10.3390/cells12182289
Awwad L, Aronheim A. Tumor Progression Reverses Cardiac Hypertrophy and Fibrosis in a Tetracycline-Regulated ATF3 Transgenic Mouse Model. Cells. 2023; 12(18):2289. https://doi.org/10.3390/cells12182289
Chicago/Turabian StyleAwwad, Lama, and Ami Aronheim. 2023. "Tumor Progression Reverses Cardiac Hypertrophy and Fibrosis in a Tetracycline-Regulated ATF3 Transgenic Mouse Model" Cells 12, no. 18: 2289. https://doi.org/10.3390/cells12182289