
Necroptosis in Organ Transplantation: Mechanisms and Potential Therapeutic Targets


Abstract
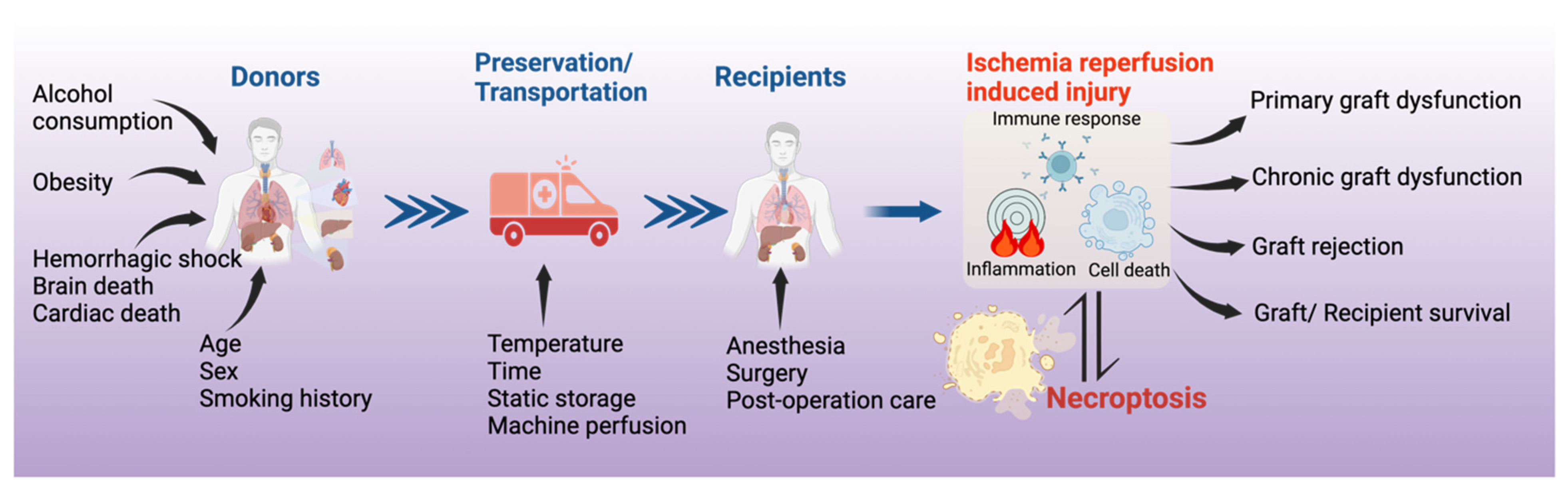
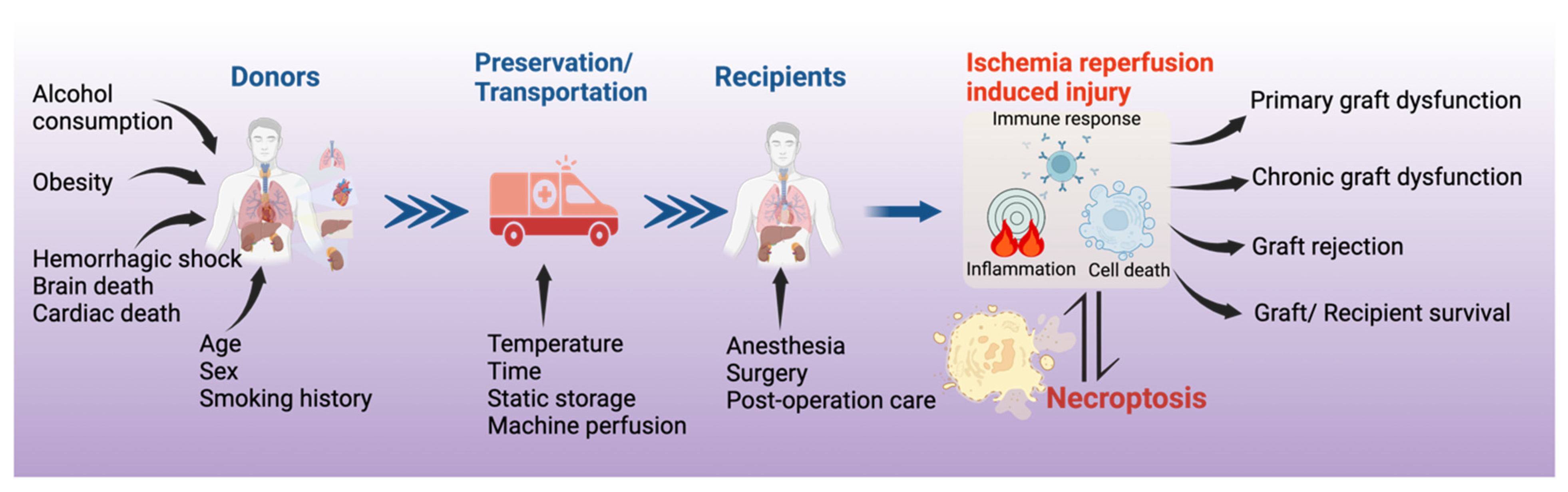
:1. Introduction
2. Materials and Methods
3. Necroptosis in Solid Organ Transplantation
3.1. Necroptosis in Kidney Transplantation
3.2. Necroptosis in Heart Transplantation
3.3. Necroptosis in Liver Transplantation
3.4. Necroptosis in Lung Transplantation
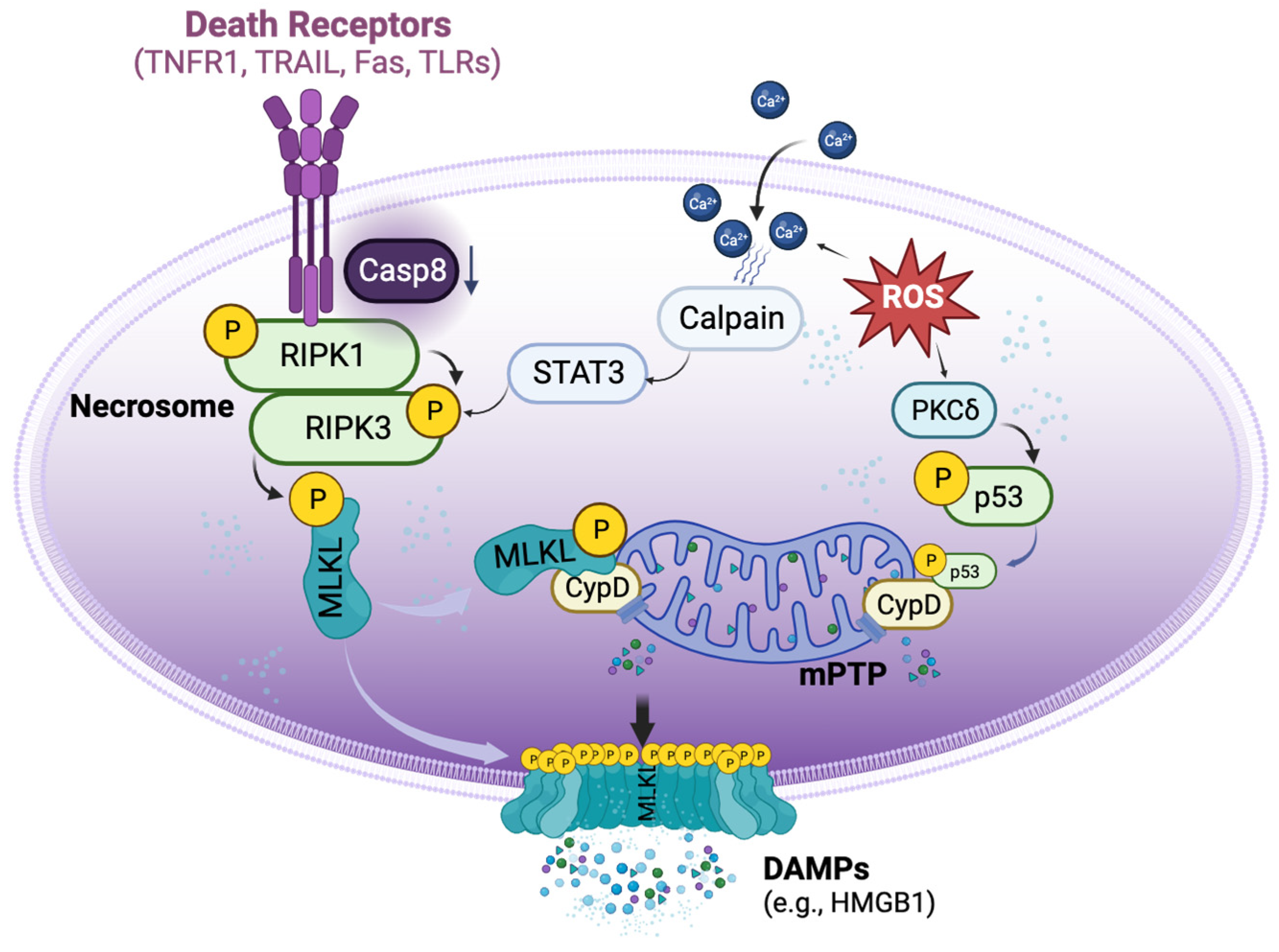
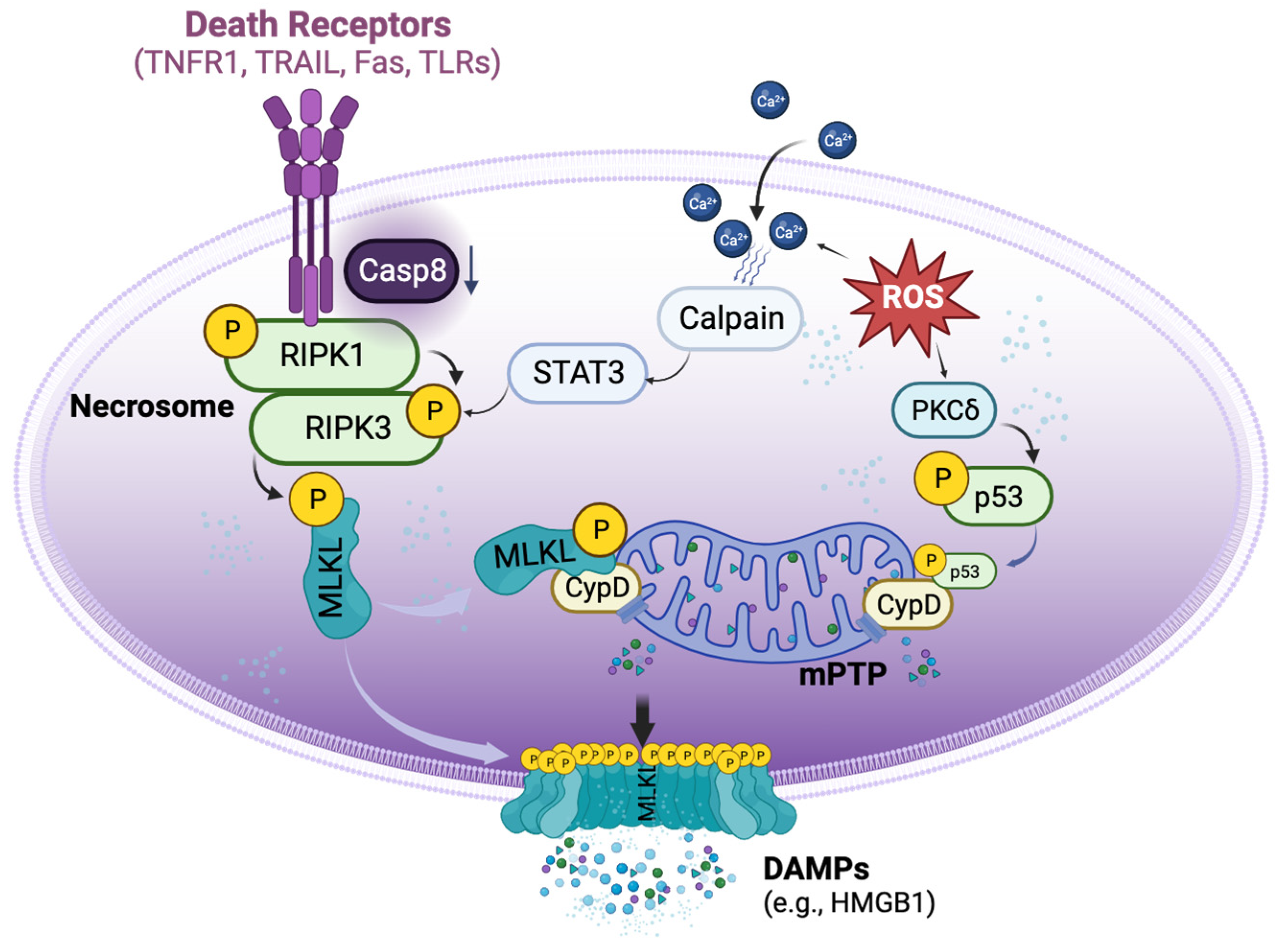
4. Mechanisms of Necroptosis in Organ Transplantation
4.1. DR-Dependent and Independent Activation of Necroptosis in Organ Transplantation
4.2. Crosstalk between Necrosome and mPTP Opening in Transplant-Related Necroptosis
4.3. Relationship between Necroptosis, Apoptosis, and Other Types of Cell Death in Organ Transplantation
5. Potential Therapeutic Targets and Substances Targeting Necroptosis
5.1. RIPK1 Inhibitors
5.2. RIPK3 Inhibitors
5.3. MLKL Inhibitor
5.4. CypD Inhibitor
5.5. Indirect Inhibitors for Necroptosis

{kind=link}
{kind=link}
| Target | Substances | Research Setting | Models | References |
| RIPK1 | Necrostatin-1, Necrostatin-1 stable | Renal/heart/lung transplant | Cell culture and animal models | [20,23,24,40,42,43,45] |
| RIPK1 | GSK2982772 | Inflammatory diseases | Phase 2a clinical trial | [66,68] |
| RIPK1 | 6E11 | Hypoxia/reoxygenation | Cell culture | [54] |
| RIPK1 | Zharp1-211 | Graft versus host disease | Animal model | [69] |
| RIPK3 | GSK′840, GSK′843, GSK′872 | Colon carcinoma | Cell culture | [70,71] |
| MLKL | Necrosulfonamide | Lung ischemia/reperfusion | Animal model | [44] |
| Cyclophilin D | Cyclosporin A | Heart transplant | Animal model | [27] |
| Protein Kinase Cδ | δV1-1 | Lung transplant | Cell culture and animal models | [39,41] |
| Calpain | N-acetyl-Leu-Leu-norleucinal | Ischemia/reperfusion | Cell culture and animal model | [40,73,74,75] |
6. Conclusions and Future Directions
6.1. Clinical Relevance of Necroptosis in Organ Transplantation
6.2. Molecular Mechanisms of Necroptosis in Organ Transplantation
6.3. Therapeutics and Biomarkers of Necroptosis in Organ Transplantation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO; Transplantation Society (TTS); Organizatión Nacional de Transplantes (ONT). Third WHO Global Consultation on Organ Donation and Transplantation: Striving to achieve self-sufficiency, March 23–25, 2010, Madrid, Spain. Transplantation 2011, 91 (Suppl. 11), S27–S28. [Google Scholar] [CrossRef]
- Lee, J.C.; Christie, J.D. Primary graft dysfunction. Proc. Am. Thorac. Soc. 2009, 6, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Al-Adhami, A.; Singh, S.S.A.; Das De, S.; Singh, R.; Panjrath, G.; Shah, A.; Dalzell, J.R.; Schroder, J.; Al-Attar, N. Primary Graft Dysfunction after Heart Transplantation—Unravelling the Enigma. Curr. Probl. Cardiol. 2022, 47, 100941. [Google Scholar] [CrossRef] [PubMed]
- Gelman, A.E.; Fisher, A.J.; Huang, H.J.; Baz, M.A.; Shaver, C.M.; Egan, T.M.; Mulligan, M.S. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction Part III: Mechanisms: A 2016 Consensus Group Statement of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2017, 36, 1114–1120. [Google Scholar] [CrossRef]
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver ischaemia–reperfusion injury: A new understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42. [Google Scholar] [CrossRef]
- Capuzzimati, M.; Hough, O.; Liu, M. Cell death and ischemia-reperfusion injury in lung transplantation. J. Heart Lung Transplant. 2022, 41, 1003–1013. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.L.; Palmer, S.M. Danger signals in regulating the immune response to solid organ transplantation. J. Clin. Investig. 2017, 127, 2464–2472. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Linkermann, A.; Bräsen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.-O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia–reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef]
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.-X.; Jevnikar, A.M. RIPK3-Mediated Necroptosis Promotes Donor Kidney Inflammatory Injury and Reduces Allograft Survival. Am. J. Transplant. 2013, 13, 2805–2818. [Google Scholar] [CrossRef]
- Jain, S.; Plenter, R.B.; Nydam, T.; Jani, A. Injury Pathways That Lead to AKI in a Mouse Kidney Transplant Model. Transplantation 2020, 104, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, Z.; Li, M.; Xiong, Y.; Wang, Y.; Peng, G.; Ye, Q. Hypothermic machine perfusion increases A20 expression which protects renal cells against ischemia/reperfusion injury by suppressing inflammation, apoptosis and necroptosis. Int. J. Mol. Med. 2016, 38, 161–171. [Google Scholar] [CrossRef]
- Pavlosky, A.; Lau, A.; Su, Y.; Lian, D.; Huang, X.; Yin, Z.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am. J. Transplant. 2014, 14, 1778–1790. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037. [Google Scholar] [CrossRef] [PubMed]
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.-X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. 2019, 19, 686–698. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W., II; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Qamar, A.; Zhao, J.; Xu, L.; McLeod, P.; Huang, X.; Jiang, J.; Liu, W.; Haig, A.; Zhang, Z.-X. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. Int. J. Mol. Sci. 2021, 22, 11038. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, X.; Mcleod, P.; Jiang, J.; Liu, W.; Haig, A.; Jevnikar, A.M.; Jiang, Z.; Zhang, Z.-X. Toll-like receptor 3 is an endogenous sensor of cell death and a potential target for induction of long-term cardiac transplant survival. Am. J. Transplant. 2021, 21, 3268–3279. [Google Scholar] [CrossRef]
- Tuuminen, R.; Holmström, E.; Raissadati, A.; Saharinen, P.; Rouvinen, E.; Krebs, R.; Lemström, K.B. Simvastatin pretreatment reduces caspase-9 and RIPK1 protein activity in rat cardiac allograft ischemia-reperfusion. Transpl. Immunol. 2016, 37, 40–45. [Google Scholar] [CrossRef]
- Rosentreter, D.; Funken, D.; Reifart, J.; Mende, K.; Rentsch, M.; Khandoga, A. RIP1-Dependent Programmed Necrosis is Negatively Regulated by Caspases During Hepatic Ischemia-Reperfusion. Shock 2015, 44, 72–76. [Google Scholar] [CrossRef]
- Ni, H.-M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.-H.; Li, T.; Ding, W.-X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)–Mixed Lineage Kinase Domain-Like Protein (MLKL)–Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McKeen, T.; Chao, X.; Chen, A.; Deng, F.; Jaeschke, H.; Ding, W.-X.; Ni, H.-M. The role of MLKL in Hepatic Ischemia-Reperfusion Injury of Alcoholic Steatotic Livers. Int. J. Biol. Sci. 2022, 18, 1096–1106. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, X.; Rao, Z.; Pan, X.; Sun, Y.; Jiang, T.; Wang, P.; Zhou, H.; Wang, X. Aging aggravated liver ischemia and reperfusion injury by promoting hepatocyte necroptosis in an endoplasmic reticulum stress-dependent manner. Ann. Transl. Med. 2020, 8, 869. [Google Scholar] [CrossRef] [PubMed]
- Olthoff, K.M.; Kulik, L.; Samstein, B.; Kaminski, M.; Abecassis, M.; Emond, J.; Shaked, A.; Christie, J.D. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transplant. 2010, 16, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Bonaccorsi-Riani, E.; Schurink, I.; Bosch, T.v.D.; Doukas, M.; Lila, K.A.; Roest, H.P.; Xhema, D.; Gianello, P.; de Jonge, J.; et al. Liver Ischemia and Reperfusion Induce Periportal Expression of Necroptosis Executor pMLKL Which Is Associated with Early Allograft Dysfunction After Transplantation. Front. Immunol. 2022, 13, 890353. [Google Scholar] [CrossRef]
- Zhao, H.; Ning, J.; Lemaire, A.; Koumpa, F.-S.; Sun, J.J.; Fung, A.; Gu, J.; Yi, B.; Lu, K.; Ma, D. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int. 2015, 87, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, Q.; Huang, H.; Suen, K.C.; Alam, A.; Cui, J.; Ciechanowicz, S.; Ning, J.; Lu, K.; Takata, M.; et al. Osteopontin mediates necroptosis in lung injury after transplantation of ischaemic renal allografts in rats. Br. J. Anaesth. 2019, 123, 519–530. [Google Scholar] [CrossRef]
- Kim, H.; Zhao, J.; Zhang, Q.; Wang, Y.; Lee, D.; Bai, X.; Turrell, L.; Chen, M.; Gao, W.; Keshavjee, S.; et al. δV1-1 Reduces Pulmonary Ischemia Reperfusion-Induced Lung Injury by Inhibiting Necrosis and Mitochondrial Localization of PKCδ and p53. Am. J. Transplant. 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Kim, H.; Zamel, R.; Bai, X.-H.; Lu, C.; Keshavjee, S.; Keshavjee, S.; Liu, M. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L595–L608. [Google Scholar] [CrossRef]
- Lee, D.; Zhao, J.; Yang, H.; Xu, S.; Kim, H.; Pacheco, S.; Keshavjee, S.; Liu, M. Effective delivery of a rationally designed intracellular peptide drug with gold nanoparticle–peptide hybrids. Nanoscale 2015, 7, 12356–12360. [Google Scholar] [CrossRef]
- Kanou, T.; Ohsumi, A.; Kim, H.; Chen, M.; Bai, X.; Guan, Z.; Hwang, D.; Cypel, M.; Keshavjee, S.; Liu, M. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1–mediated ischemia-reperfusion injury after lung transplantation. J. Heart Lung Transplant. 2018, 37, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Liang, F.; Lou, Z.; Li, Y.; Li, J.; Chen, Y.; Ding, J.; Jiang, B.; Wu, C.; Yu, H.; et al. Necrostatin-1 Alleviates Lung Ischemia-Reperfusion Injury via Inhibiting Necroptosis and Apoptosis of Lung Epithelial Cells. Cells 2022, 11, 3139. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Chen-Yoshikawa, T.F.; Tanaka, S.; Yamada, Y.; Nakajima, D.; Ohsumi, A.; Date, H. Protective effect of necrosulfonamide on rat pulmonary ischemia-reperfusion injury via inhibition of necroptosis. J. Thorac. Cardiovasc. Surg. 2022, 163, e113–e122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; O’brien, M.E.; Yu, J.; Xu, C.; Zhang, Q.; Lu, S.; Liang, L.; An, X.; McDyer, J.F.; Mallampalli, R.K. Prolonged Cold Ischemia Induces Necroptotic Cell Death in Ischemia–Reperfusion Injury and Contributes to Primary Graft Dysfunction after Lung Transplantation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 244–256. [Google Scholar] [CrossRef]
- Li, W.; Terada, Y.; Tyurina, Y.Y.; Tyurin, V.A.; Bery, A.I.; Gauthier, J.M.; Higashikubo, R.; Tong, A.Y.; Zhou, D.; Nunez-Santana, F.; et al. Necroptosis triggers spatially restricted neutrophil-mediated vascular damage during lung ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2022, 119, e2111537119. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Berghe, T.V. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020, 11, 922. [Google Scholar] [CrossRef]
- Suzuki, J.; Cole, S.E.; Batirel, S.; Kosuge, H.; Shimizu, K.; Isobe, M.; Libby, P.; Mitchell, R.N. Tumor Necrosis Factor Receptor -1 and -2 Double Deficiency Reduces Graft Arterial Disease in Murine Cardiac Allografts. Am. J. Transplant. 2003, 3, 968–976. [Google Scholar] [CrossRef]
- Gollmann-Tepeköylü, C.; Graber, M.; Pölzl, L.; Nägele, F.; Moling, R.; Esser, H.; Summerer, B.; Mellitzer, V.; Ebner, S.; Hirsch, J.; et al. Toll-like receptor 3 mediates ischaemia/reperfusion injury after cardiac transplantation. Eur. J. Cardio-Thoracic Surg. 2020, 57, 826–835. [Google Scholar] [CrossRef]
- Mallavia, B.; Liu, F.; Lefrançais, E.; Cleary, S.J.; Kwaan, N.; Tian, J.J.; Magnen, M.; Sayah, D.M.; Soong, A.; Chen, J.; et al. Mitochondrial DNA Stimulates TLR9-Dependent Neutrophil Extracellular Trap Formation in Primary Graft Dysfunction. Am. J. Respir. Cell Mol. Biol. 2020, 62, 364–372. [Google Scholar] [CrossRef]
- Li, W.; Hsiao, H.-M.; Higashikubo, R.; Saunders, B.T.; Bharat, A.; Goldstein, D.R.; Krupnick, A.S.; Gelman, A.E.; Lavine, K.J.; Kreisel, D. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. J. Clin. Investig. 2016, 1, e87315. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Delehouzé, C.; Leverrier-Penna, S.; Le Cann, F.; Comte, A.; Jacquard-Fevai, M.; Delalande, O.; Desban, N.; Baratte, B.; Gallais, I.; Faurez, F.; et al. 6E11, a highly selective inhibitor of Receptor-Interacting Protein Kinase 1, protects cells against cold hypoxia-reoxygenation injury. Sci. Rep. 2017, 7, 12931. [Google Scholar] [CrossRef]
- Bopassa, J.C.; Michel, P.; Gateau-Roesch, O.; Ovize, M.; Ferrera, R. Low-pressure reperfusion alters mitochondrial permeability transition. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2750–H2755. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B. Mitochondrial permeability transition pore—An enigmatic gatekeeper. New Horiz. Sci. Technol. 2012, 1, 47–51. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.-L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1787, 1395–1401. [Google Scholar] [CrossRef]
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial permeability transition pore-dependent necrosis. J. Mol. Cell. Cardiol. 2023, 174, 47–55. [Google Scholar] [CrossRef]
- Du, C.; Wang, S.; Diao, H.; Guan, Q.; Zhong, R.; Jevnikar, A.M. Increasing Resistance of Tubular Epithelial Cells to Apoptosis by shRNA Therapy Ameliorates Renal Ischemia-Reperfusion Injury. Am. J. Transplant. 2006, 6, 2256–2267. [Google Scholar] [CrossRef]
- Fischer, S.; Maclean, A.A.; Liu, M.; Cardella, J.A.; Slutsky, A.S.; Suga, M.; Moreira, J.F.; Keshavjee, S. Dynamic Changes in Apoptotic and Necrotic Cell Death Correlate with Severity of Ischemia-Reperfusion Injury in Lung Transplantation. Am. J. Respir. Crit. Care Med. 2000, 162, 1932–1939. [Google Scholar] [CrossRef]
- Quadri, S.M.; Segall, L.; de Perrot, M.; Han, B.; Edwards, V.; Jones, N.; Waddell, T.K.; Liu, M.; Keshavjee, S. Caspase Inhibition Improves Ischemia-Reperfusion Injury After Lung Transplantation. Am. J. Transplant. 2005, 5, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Kim, H.; Oishi, H.; Chen, M.; Iskender, I.; Sakamoto, J.; Ohsumi, A.; Guan, Z.; Hwang, D.; Waddell, T.K.; et al. Annexin V homodimer protects against ischemia reperfusion–induced acute lung injury in lung transplantation. J. Thorac. Cardiovasc. Surg. 2016, 151, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kanneganti, T.-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’En, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, e437. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Weisel, K.; Scott, N.E.; Tompson, D.J.; Votta, B.J.; Madhavan, S.; Povey, K.; Wolstenholme, A.; Simeoni, M.; Rudo, T.; Richards-Peterson, L.; et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 2017, 5, e00365. [Google Scholar] [CrossRef]
- Weisel, K.; Berger, S.; Thorn, K.; Taylor, P.C.; Peterfy, C.; Siddall, H.; Tompson, D.; Wang, S.; Quattrocchi, E.; Burriss, S.W.; et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 85. [Google Scholar] [CrossRef]
- Yu, X.; Ma, H.; Li, B.; Ji, Y.; Du, Y.; Liu, S.; Li, Z.; Hao, Y.; Tian, S.; Zhao, C.; et al. A Novel RIPK1 Inhibitor Reduces GVHD in Mice via a Non-immunosuppressive Mechanism that Restores Intestinal Homeostasis. Blood 2022, 141, 1070–1086. [Google Scholar] [CrossRef]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 Induces Apoptosis Independent of Pronecrotic Kinase Activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Zhao, H.; Jaffer, T.; Eguchi, S.; Wang, Z.; Linkermann, A.; Ma, D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015, 6, e1975. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; Marc, C.S.; Maria, K.; Suzanne, R.H. WHO Model Formulary 2008; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Bates, E.; Bode, C.; Costa, M.; Gibson, C.M.; Granger, C.; Green, C.; Grimes, K.; Harrington, R.; Huber, K.; Kleiman, N.; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008, 117, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Hernando, V.; Inserte, J.; Sartório, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell. Cardiol. 2010, 49, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Y.; Xie, L.; Zou, X.-S.; Li, N.; Yang, Y.-G.; Wu, Z.-J.; Tian, X.-Y.; Zhao, G.-Y.; Chen, M.-H. Inhibition of extracellular signal-regulated kinase/calpain-2 pathway reduces neuroinflammation and necroptosis after cerebral ischemia-reperfusion injury in a rat model of cardiac arrest. Int. Immunopharmacol. 2021, 93, 107377. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Kanou, T.; Matsui, T.; Fukui, E.; Kimura, T.; Ose, N.; Funaki, S.; Shintani, Y. Protective Effect of Calpain Inhibition During Cold Ischemia on Ischemia–reperfusion Injury After Lung Transplantation. Transplantation 2023, 107, 1945–1954. [Google Scholar] [CrossRef]
- Wong, A.; Zamel, R.; Yeung, J.; Bader, G.D.; Dos Santos, C.C.; Bai, X.; Wang, Y.; Keshavjee, S.; Liu, M. Potential therapeutic targets for lung repair during human ex vivo lung perfusion. Eur. Respir. J. 2020, 55, 1902222. [Google Scholar] [CrossRef]
- Lukac, J.; Dhaygude, K.; Saraswat, M.; Joenväärä, S.; O Syrjälä, S.; Holmström, E.J.; Krebs, R.; Renkonen, R.; I Nykänen, A.; Lemström, K.B. Plasma proteome of brain-dead organ donors predicts heart transplant outcome. J. Heart Lung Transplant. 2022, 41, 311–324. [Google Scholar] [CrossRef]
- Baciu, C.; Sage, A.; Zamel, R.; Shin, J.; Bai, X.-H.; Hough, O.; Bhat, M.; Yeung, J.C.; Cypel, M.; Keshavjee, S.; et al. Transcriptomic investigation reveals donor-specific gene signatures in human lung transplants. Eur. Respir. J. 2021, 57, 2000327. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y.; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef]
- Zheng, P.; Kang, J.; Xing, E.; Zheng, B.; Wang, X.; Zhou, H. Lung Inflation with Hydrogen During the Cold Ischemia Phase Alleviates Lung Ischemia-Reperfusion Injury by Inhibiting Pyroptosis in Rats. Front. Physiol. 2021, 12, 699344. [Google Scholar] [CrossRef]
- Lu, J.; Xu, L.; Zeng, Z.; Xue, C.; Li, J.; Chen, X.; Zhou, P.; Lin, S.; Liao, Y.; Du, X.; et al. Normothermic ex vivo Heart Perfusion Combined with Melatonin Enhances Myocardial Protection in Rat Donation After Circulatory Death Hearts via Inhibiting NLRP3 Inflammasome-Mediated Pyroptosis. Front. Cell Dev. Biol. 2021, 9, 733183. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Main, K.; Aujla, T.; Keshavjee, S.; Liu, M. Necroptosis in Organ Transplantation: Mechanisms and Potential Therapeutic Targets. Cells 2023, 12, 2296. https://doi.org/10.3390/cells12182296
Zhao Y, Main K, Aujla T, Keshavjee S, Liu M. Necroptosis in Organ Transplantation: Mechanisms and Potential Therapeutic Targets. Cells. 2023; 12(18):2296. https://doi.org/10.3390/cells12182296
Chicago/Turabian StyleZhao, Yajin, Kimberly Main, Tanroop Aujla, Shaf Keshavjee, and Mingyao Liu. 2023. "Necroptosis in Organ Transplantation: Mechanisms and Potential Therapeutic Targets" Cells 12, no. 18: 2296. https://doi.org/10.3390/cells12182296
APA StyleZhao, Y., Main, K., Aujla, T., Keshavjee, S., & Liu, M. (2023). Necroptosis in Organ Transplantation: Mechanisms and Potential Therapeutic Targets. Cells, 12(18), 2296. https://doi.org/10.3390/cells12182296