
Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS
 ,
,
Abstract
:1. Introduction
2. Absence of Primary Neurological Disease in HGPS
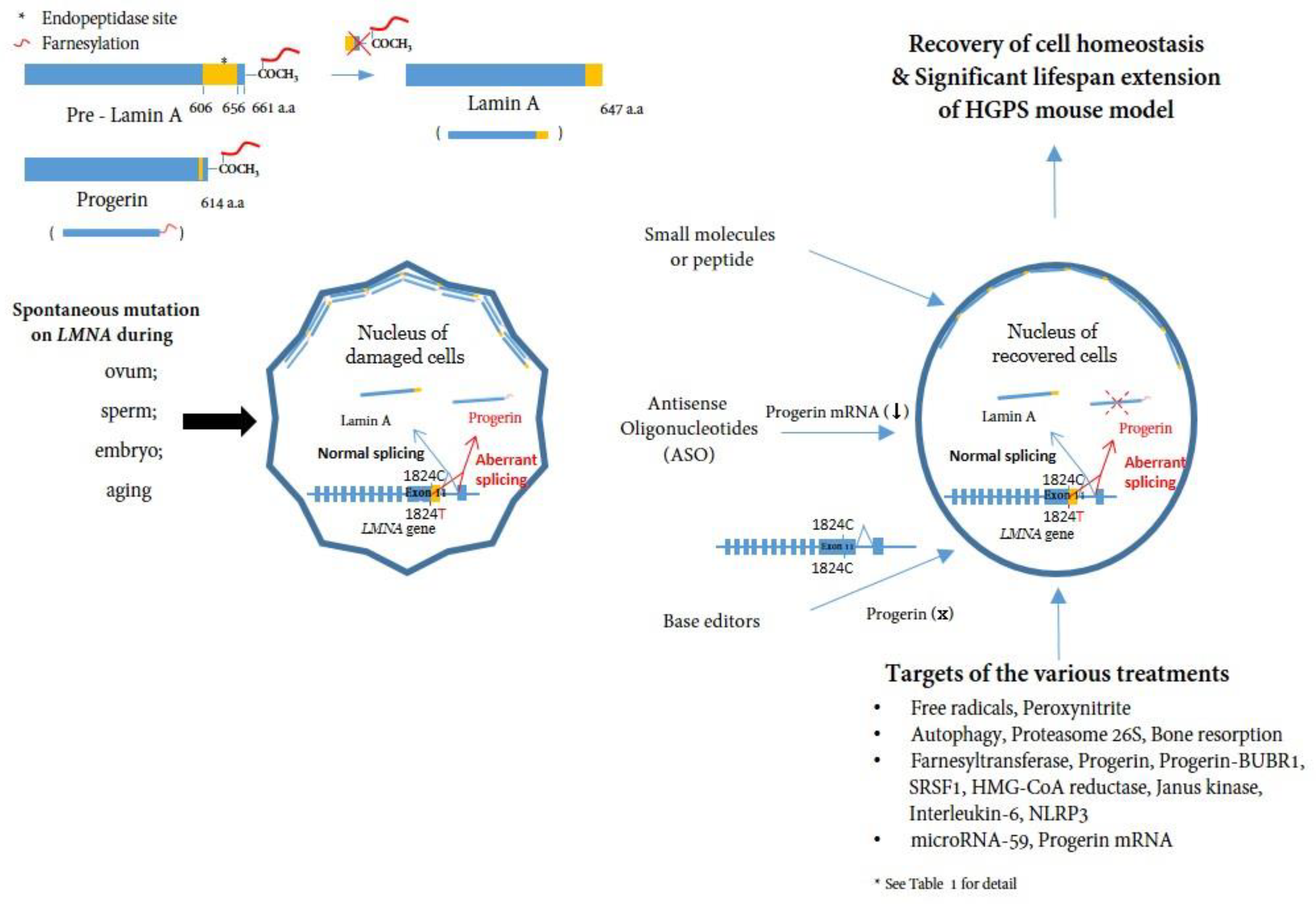
3. Dysfunctional Progerin Expression
- Effect of progerin at the cellular level
- b.
- Systemic effect of progerin or effect of progerin on tissues
4. Targeting and Inhibiting Progerin at mRNA and DNA Level
5. Treatments and Clinical Trials for HGPS

{kind=link}
| Treatments | Targets | Action | Study Design (Cell or Animal Model) | Refs |
|---|---|---|---|---|
| Rapamycin | mTOR | Autophagic induction | HGPS fibroblasts | [33,110] |
| Sulforaphane (SFN) | Free radicals | SFN (antioxidant derived from cruciferous vegetables) stimulates proteasome activity and autophagy in normal and HGPS fibroblast cultures | HGPS fibroblasts, Human nucleus pulposus cells, LmnaG609G/G609G mice | [112,113] |
| Metformin | serine/arginine-rich splicing factor 1 (SRSF1) | A-type lamins is controlled by SRSF1. SRSF1 expression is transcriptionally regulated by the antidiabetic drug metformin. | HGPS mesenchymal stem cells, HGPS fibroblasts, LmnaG609G/G609G fibroblasts | [114] |
| Pravastatin and Zoledronic acid | hydroxymethylglutaryl-CoA (HMG-CoA) reductase /bone resorption | Zoledronic acid prevents bone fractures Pravastatin is used for treating high cholesterol and preventing heart attacks and strokes | Transgenic G608G BAC mice | [89] |
| Neuropeptide Y | Autophagy | NPY mediates caloric restriction-induced autophagy | HGPS fibroblasts | [111] |
| MG132 | Proteasome 26S | Autophagic activation after the loss of proteasomal activity. Downregulation of SRSF1 and reduction of inflammatory cytokines | HGPS fibroblasts, LmnaG609G/G609G mouse, HGPS-like fibroblasts, MAD-B syndrome fibroblasts | [115,116] |
| Unique Progerin C-terminal peptide (UPCP) | Progerin-BUBR1 | UPCP blocks the binding between Progerin and BUBR1 | HGPS fibroblasts, LmnaG609G/G609G mouse | [117] |
| Lonafarnib | Farnesyltransferase | Non-farnesylated prelamin A production by inhibition of farnesyltransferase activity | HGPS fibroblast, Zmpste24−/− fibroblast, Transgenic G608G BAC mice Zmpste24−/− mice | [119,127,128,129] |
| Progerinin (SLC-D011) | Progerin | The small molecule specifically bindsto Progerin and induces its degradation | HGPS fibroblasts, WRN fibroblasts and cardiomyocytes, LMNAG609G/G609G mice | [59,96,124] |
| MnTBAP and Baricitinib | Peroxynitrite /Janus kinase | Inhibition of peroxynitrite- induced oxidative reactions /anti-inflammatory action by JAK1/2 inhibitor | HGPS fibroblasts | [118] |
| Tocilizumab | IL-6 | Immunosuppression by anti-IL-6 antibody | HGPS fibroblasts, LmnaG609G/G609G mouse | [120] |
| Baricitinib and lonafarnib | Janus kinase /Farnesyltransferase | Inhibition of peroxynitrite-induced oxidative reactions /anti-inflammatory action by JAK1/2 inhibitor | HGPS fibroblasts, FPLD2 syndrome fibroblasts, MAD-B syndrome fibroblasts | [125] |
| Anti-miR-59 | microRNA-59 | AAV9-mediated miR-59 inhibition by antisense oligonucleotide | HGPS fibroblasts, LmnaG609G/G609G mouse | [53] |
| MCC950 | NLRP3 | Inflammasome inactivation by NLRP3 inhibition | HGPS fibroblast, Zmpste24−/− mice, LMNAG609G/G609G mice | [95] |
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gilford, H. Progeria: A form of senilism. Practitioner 1904, 73, 188–217. [Google Scholar]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef]
- Burke, B.; Stewart, C.L. Life at the edge: The nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 575–585. [Google Scholar] [CrossRef]
- Kipling, D.; Davis, T.; Ostler, E.L.; Faragher, R.G. What can progeroid syndromes tell us about human aging? Science 2004, 305, 1426–1431. [Google Scholar] [CrossRef]
- Miller, R.A. “Accelerated aging”: A primrose path to insight? Aging Cell 2004, 3, 47–51. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Gordon, L.B.; Rothman, F.G.; López-Otín, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford progeria syndrome: A premature aging disease. Mol. Neurobiol. 2018, 55, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin a and nuclear lamin c. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Wydner, K.L.; McNeil, J.A.; Lin, F.; Worman, H.J.; Lawrence, J.B. Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization. Genomics 1996, 32, 474–478. [Google Scholar] [CrossRef]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. CDNA sequencing of nuclear lamins a and c reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Burke, B.; Stewart, C.L. The a-type lamins: Nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc. Med. 2001, 11, 280–285. [Google Scholar] [CrossRef]
- Davies, B.S.; Fong, L.G.; Yang, S.H.; Coffinier, C.; Young, S.G. The posttranslational processing of prelamin a and disease. Annu. Rev. Genom. Hum. Genet. 2009, 10, 153–174. [Google Scholar] [CrossRef]
- Coffinier, C.; Jung, H.J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J.; et al. Direct synthesis of lamin a, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 2010, 285, 20818–20826. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson-Gilford progeria mutant lamin a primarily targets human vascular cells as detected by an anti-lamin a g608g antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.; Shanker, V.; Sharma, N. Hutchinson-Gilford progeria syndrome: A rare case report. Indian Dermatol. Online J. 2014, 5, 478–481. [Google Scholar] [CrossRef]
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific mir-9 microRNA. Cell Rep. 2012, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Jung, H.J.; Lee, J.M.; Fong, L.G. Nuclear lamins and neurobiology. Mol. Cell Biol. 2014, 34, 2776–2785. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin a but not lamin c by mir-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef]
- Jung, H.J.; Tu, Y.; Yang, S.H.; Tatar, A.; Nobumori, C.; Wu, D.; Young, S.G.; Fong, L.G. New LMNA knock-in mice provide a molecular mechanism for the ‘segmental aging’ in Hutchinson-Gilford progeria syndrome. Hum. Mol. Genet. 2014, 23, 1506–1515. [Google Scholar] [CrossRef]
- Coffinier, C.; Chang, S.Y.; Nobumori, C.; Tu, Y.; Farber, E.A.; Toth, J.I.; Fong, L.G.; Young, S.G. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin b2 deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 5076–5081. [Google Scholar] [CrossRef]
- Coffinier, C.; Fong, L.G.; Young, S.G. Lincing lamin b2 to neuronal migration: Growing evidence for cell-specific roles of b-type lamins. Nucleus 2010, 1, 407–411. [Google Scholar] [CrossRef]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in lamin b1 and lamin b2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Fu, Y.H. Autosomal dominant leukodystrophy caused by lamin b1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 2010, 98, 337–357. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptácek, L.J.; Fu, Y.H. Lamin b1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin b1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef]
- Vidak, S.; Kubben, N.; Dechat, T.; Foisner, R. Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2α (LAP2α) through expression of extracellular matrix proteins. Genes Dev. 2015, 29, 2022–2036. [Google Scholar] [CrossRef]
- Gordon, L.B.; Norris, W.; Hamren, S.; Goodson, R.; LeClair, J.; Massaro, J.; Lyass, A.; D’Agostino, R.B., Sr.; Tuminelli, K.; Kieran, M.W.; et al. Plasma Progerin in patients with Hutchinson-Gilford progeria syndrome: Immunoassay development and clinical evaluation. Circulation 2023, 147, 1734–1744. [Google Scholar] [CrossRef]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant nrf2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef]
- Kychygina, A.; Dall’Osto, M.; Allen, J.A.M.; Cadoret, J.C.; Piras, V.; Pickett, H.A.; Crabbe, L. Progerin impairs 3-d genome organization and induces fragile telomeres by limiting the DNTP pools. Sci. Rep. 2021, 11, 13195. [Google Scholar] [CrossRef]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of proliferative defects associated with processing-defective lamin a mutants by htert or inactivation of p53. Mol. Biol. Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of Progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.; Lim, J.S.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces lap2α-telomere association in Hutchinson-Gilford progeria. eLife 2015, 4, e07759. [Google Scholar] [CrossRef] [PubMed]
- Noda, A.; Mishima, S.; Hirai, Y.; Hamasaki, K.; Landes, R.D.; Mitani, H.; Haga, K.; Kiyono, T.; Nakamura, N.; Kodama, Y. Progerin, the protein responsible for the Hutchinson-Gilford progeria syndrome, increases the unrepaired DNA damages following exposure to ionizing radiation. Genes Environ. 2015, 37, 13. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, A.; Ong, P.F.; Foo, M.X.R.; Liebl, D.; Hor, L.P.; Stewart, C.L.; Dreesen, O. Heterochromatin loss as a determinant of Progerin-induced DNA damage in Hutchinson-Gilford progeria. Aging Cell 2020, 19, e13108. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Stiber, J.A.; Rosenberg, P.B. The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium 2011, 49, 341–349. [Google Scholar] [CrossRef]
- Wang, W.P.; Wang, J.Y.; Lin, W.H.; Kao, C.H.; Hung, M.C.; Teng, Y.C.; Tsai, T.F.; Chi, Y.H. Progerin in muscle leads to thermogenic and metabolic defects via impaired calcium homeostasis. Aging Cell 2020, 19, e13090. [Google Scholar] [CrossRef]
- Luo, X.; Jiang, X.; Li, J.; Bai, Y.; Li, Z.; Wei, P.; Sun, S.; Liang, Y.; Han, S.; Li, X.; et al. Insulin-like growth factor-1 attenuates oxidative stress-induced hepatocyte premature senescence in liver fibrogenesis via regulating nuclear p53-Progerin interaction. Cell Death Dis. 2019, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Bai, Y.; He, S.; Sun, S.; Jiang, X.; Yang, Z.; Lu, D.; Wei, P.; Liang, Y.; Peng, C.; et al. Sirtuin 1 ameliorates defenestration in hepatic sinusoidal endothelial cells during liver fibrosis via inhibiting stress-induced premature senescence. Cell Prolif. 2021, 54, e12991. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, X.; Zhang, Y.; Gao, Z.; Liu, Y.; Hu, J.; Hu, X.; Li, L.; Shi, J.; Gao, N. Nuclear accumulation of ubc9 contributes to SUMOylation of lamin a/c and nucleophagy in response to DNA damage. J. Exp. Clin. Cancer Res. 2019, 38, 67. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Djabali, K. Autophagic removal of farnesylated carboxy-terminal lamin peptides. Cells 2018, 7, 33. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, J.; Jiang, X.; Li, X.; Zhang, B.; Luo, X. Nucleophagic degradation of Progerin ameliorates defenestration in liver sinusoidal endothelium due to sirt1-mediated deacetylation of nuclear lc3. Cells 2022, 11, 3918. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, J.; Jeong, S.; Jo, I.; Kang, S.M.; Park, B.J.; Ha, N.C. Structural basis for the interaction between unfarnesylated progerin and the IG-like domain of lamin a/c in premature aging disorders. Biochem. Biophys. Res. Commun. 2022, 637, 210–217. [Google Scholar] [CrossRef]
- Frankel, D.; Delecourt, V.; Novoa-Del-Toro, E.M.; Robin, J.D.; Airault, C.; Bartoli, C.; Carabalona, A.; Perrin, S.; Mazaleyrat, K.; De Sandre-Giovannoli, A.; et al. Mir-376a-3p and mir-376b-3p overexpression in Hutchinson-Gilford progeria fibroblasts inhibits cell proliferation and induces premature senescence. iScience 2022, 25, 103757. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, N.; Sui, T.; Li, G.; Wang, Z.; Liu, M.; Zhu, X.; Huang, B.; Lu, J.; Li, Z.; et al. Anti-hsa-mir-59 alleviates premature senescence associated with Hutchinson-Gilford progeria syndrome in mice. EMBO J. 2023, 42, e110937. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin a-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Skoczyńska, A.; Budzisz, E.; Dana, A.; Rotsztejn, H. New look at the role of Progerin in skin aging. Prz. Menopauzalny 2015, 14, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of Progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech. Ageing Dev. 2010, 131, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin a-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef]
- Cao, K.; Capell, B.C.; Erdos, M.R.; Djabali, K.; Collins, F.S. A lamin a protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4949–4954. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Jung, Y.S.; Yoon, M.H.; Kang, S.M.; Oh, A.Y.; Lee, J.H.; Jun, S.Y.; Woo, T.G.; Chun, H.Y.; Kim, S.K.; et al. Interruption of Progerin-lamin a/c binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Investig. 2016, 126, 3879–3893. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin a that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arter. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef]
- Leung, G.K.; Schmidt, W.K.; Bergo, M.O.; Gavino, B.; Wong, D.H.; Tam, A.; Ashby, M.N.; Michaelis, S.; Young, S.G. Biochemical studies of zmpste24-deficient mice. J. Biol. Chem. 2001, 276, 29051–29058. [Google Scholar] [CrossRef] [PubMed]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective prelamin a processing and muscular and adipocyte alterations in zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andrés-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; López-Otín, C.; Andrés, V. Vascular smooth muscle-specific Progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef]
- Nevado, R.M.; Hamczyk, M.R.; Gonzalo, P.; Andrés-Manzano, M.J.; Andrés, V. Premature vascular aging with features of plaque vulnerability in an atheroprone mouse model of Hutchinson-Gilford progeria syndrome with LDLR deficiency. Cells 2020, 9, 2252. [Google Scholar] [CrossRef]
- Del Campo, L.; Sánchez-López, A.; González-Gómez, C.; Andrés-Manzano, M.J.; Dorado, B.; Andrés, V. Vascular smooth muscle cell-specific progerin expression provokes contractile impairment in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated by nitrite treatment. Cells 2020, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, L.; Sánchez-López, A.; Salaices, M.; von Kleeck, R.A.; Expósito, E.; González-Gómez, C.; Cussó, L.; Guzmán-Martínez, G.; Ruiz-Cabello, J.; Desco, M.; et al. Vascular smooth muscle cell-specific progerin expression in a mouse model of Hutchinson-Gilford progeria syndrome promotes arterial stiffness: Therapeutic effect of dietary nitrite. Aging Cell 2019, 18, e12936. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Qin, W.; Tang, X.; Meng, Y.; Hu, W.; Zhang, S.; Qian, M.; Liu, Z.; Cao, X.; Pang, Q.; et al. Vascular endothelium-targeted sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci. Adv. 2020, 6, eaay5556. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Dorado, B.; Andrés, V. Molecular and cellular mechanisms driving cardiovascular disease in Hutchinson-Gilford progeria syndrome: Lessons learned from animal models. Cells 2021, 10, 1157. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [CrossRef]
- Stehbens, W.E.; Wakefield, S.J.; Gilbert-Barness, E.; Olson, R.E.; Ackerman, J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc. Pathol. 1999, 8, 29–39. [Google Scholar] [CrossRef]
- Stehbens, W.E.; Delahunt, B.; Shozawa, T.; Gilbert-Barness, E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc. Pathol. 2001, 10, 133–136. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andrés-Manzano, M.J.; Misteli, T.; López-Otín, C.; Andrés, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736. [Google Scholar] [CrossRef]
- Kinoshita, D.; Nagasawa, A.; Shimizu, I.; Ito, T.K.; Yoshida, Y.; Tsuchida, M.; Iwama, A.; Hayano, T.; Minamino, T. Progerin impairs vascular smooth muscle cell growth via the DNA damage response pathway. Oncotarget 2017, 8, 34045–34056. [Google Scholar] [CrossRef]
- Jiang, Y.; Ji, J.Y. Progerin-induced impairment in wound healing and proliferation in vascular endothelial cells. Front. Aging 2022, 3, 844885. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; Mahajan, U.; Shashkova, E.V.; Lin, C.J.; Mecham, R.P.; Gonzalo, S. Progerin induces a phenotypic switch in vascular smooth muscle cells and triggers replication stress and an aging-associated secretory signature. GeroScience 2023, 45, 965–982. [Google Scholar] [CrossRef]
- von Kleeck, R.; Castagnino, P.; Assoian, R.K. Progerin mislocalizes myocardin-related transcription factor in Hutchinson-guilford progeria syndrome. Vasc. Biol. 2022, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, A.; Espinós-Estévez, C.; González-Gómez, C.; Gonzalo, P.; Andrés-Manzano, M.J.; Fanjul, V.; Riquelme-Borja, R.; Hamczyk, M.R.; Macías, Á.; Del Campo, L.; et al. Cardiovascular Progerin suppression and lamin a restoration rescue Hutchinson-Gilford progeria syndrome. Circulation 2021, 144, 1777–1794. [Google Scholar] [CrossRef] [PubMed]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin a inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef]
- Bidault, G.; Vatier, C.; Capeau, J.; Vigouroux, C.; Béréziat, V. Lmna-linked lipodystrophies: From altered fat distribution to cellular alterations. Biochem. Soc. Trans. 2011, 39, 1752–1757. [Google Scholar] [CrossRef] [PubMed]
- Najdi, F.; Krüger, P.; Djabali, K. Impact of progerin expression on adipogenesis in Hutchinson-Gilford progeria skin-derived precursor cells. Cells 2021, 10, 1598. [Google Scholar] [CrossRef]
- Xiong, Z.M.; LaDana, C.; Wu, D.; Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from IPS cells. Aging 2013, 5, 288–303. [Google Scholar] [CrossRef]
- Mateos, J.; Landeira-Abia, A.; Fafián-Labora, J.A.; Fernández-Pernas, P.; Lesende-Rodríguez, I.; Fernández-Puente, P.; Fernández-Moreno, M.; Delmiro, A.; Martín, M.A.; Blanco, F.J.; et al. Itraq-based analysis of Progerin expression reveals mitochondrial dysfunction, reactive oxygen species accumulation and altered proteostasis. Stem Cell Res. Ther. 2015, 6, 119. [Google Scholar] [CrossRef]
- Gordon, C.M.; Gordon, L.B.; Snyder, B.D.; Nazarian, A.; Quinn, N.; Huh, S.; Giobbie-Hurder, A.; Neuberg, D.; Cleveland, R.; Kleinman, M.; et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Miner. Res. 2011, 26, 1670–1679. [Google Scholar] [CrossRef]
- Cubria, M.B.; Suarez, S.; Masoudi, A.; Oftadeh, R.; Kamalapathy, P.; DuBose, A.; Erdos, M.R.; Cabral, W.A.; Karim, L.; Collins, F.S.; et al. Evaluation of musculoskeletal phenotype of the g608g progeria mouse model with lonafarnib, pravastatin, and zoledronic acid as treatment groups. Proc. Natl. Acad. Sci. USA 2020, 117, 12029–12040. [Google Scholar] [CrossRef]
- Gargiuli, C.; Schena, E.; Mattioli, E.; Columbaro, M.; D’Apice, M.R.; Novelli, G.; Greggi, T.; Lattanzi, G. Lamins and bone disorders: Current understanding and perspectives. Oncotarget 2018, 9, 22817–22831. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [PubMed]
- Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Béréziat, V. Progerin expression induces inflammation, oxidative stress and senescence in human coronary endothelial cells. Cells 2020, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by Progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef]
- Messner, M.; Ghadge, S.K.; Maurer, T.; Graber, M.; Staggl, S.; Christine Maier, S.; Pölzl, G.; Zaruba, M.M. ZMPSTE24 is associated with elevated inflammation and Progerin mRNA. Cells 2020, 9, 1981. [Google Scholar] [CrossRef]
- González-Dominguez, A.; Montañez, R.; Castejón-Vega, B.; Nuñez-Vasco, J.; Lendines-Cordero, D.; Wang, C.; Mbalaviele, G.; Navarro-Pando, J.M.; Alcocer-Gómez, E.; Cordero, M.D. Inhibition of the NLRP3 inflammasome improves lifespan in animal murine model of Hutchinson-Gilford progeria. EMBO Mol. Med. 2021, 13, e14012. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Lee, S.J.; Ahn, J.; Yi, S.A.; Nam, K.H.; Park, S.; Woo, T.G.; Cho, J.H.; Lee, J.; et al. Human WRN is an intrinsic inhibitor of Progerin, abnormal splicing product of lamin a. Sci. Rep. 2021, 11, 9122. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Libina, N.; Janes, J.; Martin, G.M.; Campisi, J.; Oshima, J. Correction of cellular phenotypes of Hutchinson-Gilford progeria cells by RNA interference. Hum. Genet. 2005, 118, 444–450. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of lmna splicing as a strategy to treat prelamin a diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Nielsen, M.W.; Stage, M.H.; Arnspang, E.C. Nuclear envelope morphology change upon repetitive treatment with modified antisense oligonucleotides targeting Hutchinson-Gilford progeria syndrome. Biochem. Biophys. Rep. 2023, 33, 101411. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Lévy, N.; De Sandre-Giovannoli, A. Antisense-based Progerin downregulation in HGPS-like patients’ cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Coté, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin a and lamin a appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Puttaraju, M.; Jackson, M.; Klein, S.; Shilo, A.; Bennett, C.F.; Gordon, L.; Rigo, F.; Misteli, T. Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Erdos, M.R.; Cabral, W.A.; Tavarez, U.L.; Cao, K.; Gvozdenovic-Jeremic, J.; Narisu, N.; Zerfas, P.M.; Crumley, S.; Boku, Y.; Hanson, G.; et al. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. The challenges and strategies of antisense oligonucleotide drug delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Whisenant, D.; Lim, K.; Revêchon, G.; Yao, H.; Bergo, M.O.; Machtel, P.; Kim, J.S.; Eriksson, M. Transient expression of an adenine base editor corrects the Hutchinson-Gilford progeria syndrome mutation and improves the skin phenotype in mice. Nat. Commun. 2022, 13, 3068. [Google Scholar] [CrossRef]
- Clements, C.S.; Bikkul, M.U.; Ofosu, W.; Eskiw, C.; Tree, D.; Makarov, E.; Kill, I.R.; Bridger, J.M. Presence and distribution of Progerin in HGPS cells is ameliorated by drugs that impact on the mevalonate and mtor pathways. Biogerontology 2019, 20, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Ferreira-Marques, M.; Cortes, L.; Valero, J.; Pereira, D.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide y enhances Progerin clearance and ameliorates the senescent phenotype of human Hutchinson-Gilford progeria syndrome cells. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances Progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, D.; Zheng, C.; Gao, B.; Fan, J.; Cheng, P.; Liu, B.; Yang, L.; Luo, Z. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics 2019, 9, 2252–2267. [Google Scholar] [CrossRef]
- Egesipe, A.L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.L.; Navarro, C.; Sandre-Giovannoli, A.; Levy, N.; Peschanski, M.; Nissan, X. Metformin decreases Progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech. Dis. 2016, 2, 16026. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Lévy, N. MG132-induced Progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Harhouri, K.; Cau, P.; Casey, F.; Guedenon, K.M.; Doubaj, Y.; Van Maldergem, L.; Mejia-Baltodano, G.; Bartoli, C.; De Sandre-Giovannoli, A.; Lévy, N. MG132 induces Progerin clearance and improves disease phenotypes in HGPS-like patients’ cells. Cells 2022, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Hu, Q.; Sui, T.; Fu, L.; Zhang, X.; Wang, Y.; Zhu, X.; Huang, B.; Lu, J.; Li, Z. Unique Progerin C-terminal peptide ameliorates Hutchinson-Gilford progeria syndrome phenotype by rescuing bubr1. Nat. Aging 2023, 3, 185–201. [Google Scholar] [CrossRef]
- Vehns, E.; Arnold, R.; Djabali, K. Impact of MnTBAP and baricitinib treatment on Hutchinson-Gilford progeria fibroblasts. Pharmaceuticals 2022, 15, 945. [Google Scholar] [CrossRef]
- Monterrubio-Ledezma, F.; Navarro-García, F.; Massieu, L.; Mondragón-Flores, R.; Soto-Ponce, L.A.; Magaña, J.J.; Cisneros, B. Rescue of Mitochondrial Function in Hutchinson-Gilford Progeria Syndrome by the Pharmacological Modulation of Exportin CRM1. Cells 2023, 12, 275. [Google Scholar] [CrossRef]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef]
- Schmidt, E.; Nilsson, O.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J. Biol. Chem. 2012, 287, 33512–33522. [Google Scholar] [CrossRef] [PubMed]
- Strandgren, C.; Nasser, H.A.; McKenna, T.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015, 29, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Revêchon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtačnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Ahn, J.; Kim, J.E.; Kim, S.Y.; Kang, S.Y.; Joo, J.; Park, S.; Cho, J.H.; Woo, T.G.; et al. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun. Biol. 2021, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, R.; Lederer, E.M.; Schena, E.; Lattanzi, G.; Djabali, K. Impact of combined baricitinib and FTI treatment on adipogenesis in Hutchinson-Gilford progeria syndrome and other lipodystrophic laminopathies. Cells 2023, 12, 1350. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, Y.; Kim, S.; Chang, Y.; Kim, Y.; Kwon, Y.; Kim, J. Transcriptional activation of endogenous oct4 via the crispr/dcas9 activator ameliorates Hutchinson-Gilford progeria syndrome in mice. Aging Cell 2023, 22, e13825. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of Progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 2005, 102, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Olive, M.; Erdos, M.R.; Cao, K.; Faddah, D.A.; Tavarez, U.L.; Conneely, K.N.; Qu, X.; San, H.; Ganesh, S.K.; et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 15902–15907. [Google Scholar] [CrossRef]
- Dhillon, S. Lonafarnib: First approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.G.; Frost, D.; Meta, M.; Qiao, X.; Yang, S.H.; Coffinier, C.; Young, S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006, 311, 1621–1623. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 2011, 108, 4997–5002. [Google Scholar] [CrossRef] [PubMed]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 2016, 7, e2105. [Google Scholar] [CrossRef] [PubMed]
- Basso, A.D.; Kirschmeier, P.; Bishop, W.R. Lipid posttranslational modifications. Farnesyl transferase inhibitors Thematic Review Series. J. Lipid Res. 2006, 47, 15–31. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef]
- Chen, X.; Yao, H.; Kashif, M.; Revêchon, G.; Eriksson, M.; Hu, J.; Wang, T.; Liu, Y.; Tüksammel, E.; Strömblad, S.; et al. A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 2021, 10, e63284. [Google Scholar] [CrossRef]
- Cabral, W.A.; Tavarez, U.L.; Beeram, I.; Yeritsyan, D.; Boku, Y.D.; Eckhaus, M.A.; Nazarian, A.; Erdos, M.R.; Collins, F.S. Genetic reduction of mtor extends lifespan in a mouse model of Hutchinson-Gilford progeria syndrome. Aging Cell 2021, 20, e13457. [Google Scholar] [CrossRef]
- Abutaleb, N.O.; Atchison, L.; Choi, L.; Bedapudi, A.; Shores, K.; Gete, Y.; Cao, K.; Truskey, G.A. Lonafarnib and everolimus reduce pathology in IPSC-derived tissue engineered blood vessel model of Hutchinson-Gilford progeria syndrome. Sci. Rep. 2023, 13, 5032. [Google Scholar] [CrossRef]
| Clinical Trials NCT# | Drugs | Stage | Number of Individuals | Study Type | First Posted (Year) /Recruiting Status | Status or Main Finding |
|---|---|---|---|---|---|---|
| NCT00425607 | Lonafarnib | Phase II | 29 | Interventional | 2007 /complete | Found life span extension (about 1.6 years) |
| NCT00731016 | Zoledronate and pravastatin | Phase II | 15 | Interventional | 2008 /complete | Found the reduction of the alternative prenylation induced by Lonafarnib |
| NCT00879034 | Zoledronate, pravastatin, and Lonafarnib | Phase II | 5 | Interventional | 2009 /complete | No additional improvement of the tri-therapy as compared to lonafarnib alone |
| NCT00916747 | Zoledronate, pravastatin, and Lonafarnib | Phase II | 85 | Interventional | 2009 /active | Optimizing the efficient tri-therapy combination |
| NCT02579044 | Everolimus and Lonafarnib | Phase I/II | 80 | Interventional | 2015 /Enrolling by invitation | Defining Maximum-tolerated dose (MTD) of everolimus & efficacy of combination |
| NCT04512963 | Progerinin | Phase I | 64 | Interventional | 2020 /complete | No safety concerns with the Progerinin at all testing doses and food conditions (up to the maximum dose of 2400 mg) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-H.; Chung, Y.-H.; Woo, T.-G.; Kang, S.-M.; Park, S.; Park, B.-J. Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS. Cells 2023, 12, 2299. https://doi.org/10.3390/cells12182299
Kim B-H, Chung Y-H, Woo T-G, Kang S-M, Park S, Park B-J. Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS. Cells. 2023; 12(18):2299. https://doi.org/10.3390/cells12182299
Chicago/Turabian StyleKim, Bae-Hoon, Yeon-Ho Chung, Tae-Gyun Woo, So-Mi Kang, Soyoung Park, and Bum-Joon Park. 2023. "Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS" Cells 12, no. 18: 2299. https://doi.org/10.3390/cells12182299
APA StyleKim, B.-H., Chung, Y.-H., Woo, T.-G., Kang, S.-M., Park, S., & Park, B.-J. (2023). Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS. Cells, 12(18), 2299. https://doi.org/10.3390/cells12182299