
Emerging Roles of Ubiquitination in Biomolecular Condensates

Abstract
:1. Introduction
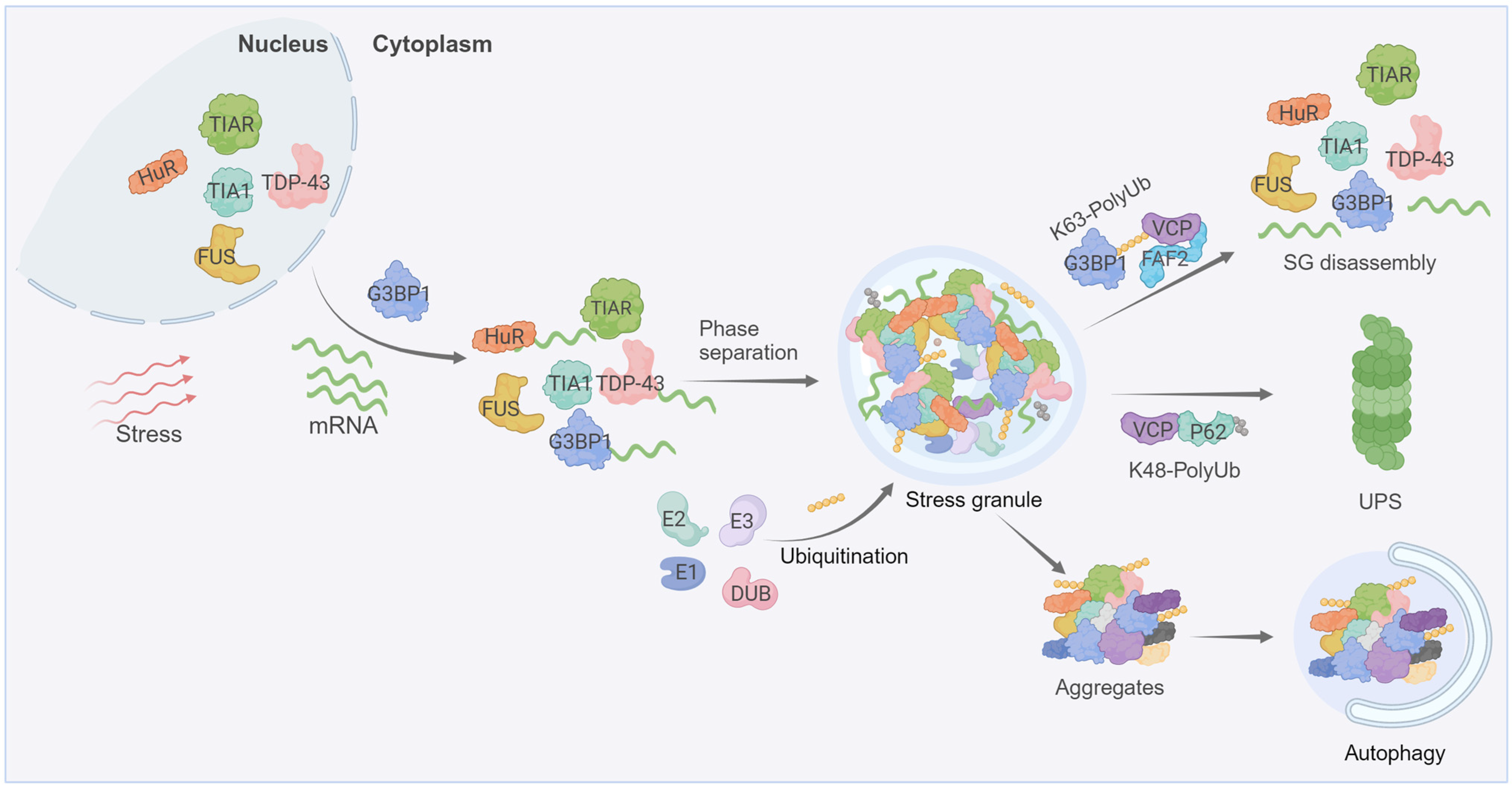
2. Ubiquitin in Stress Granule Dynamics
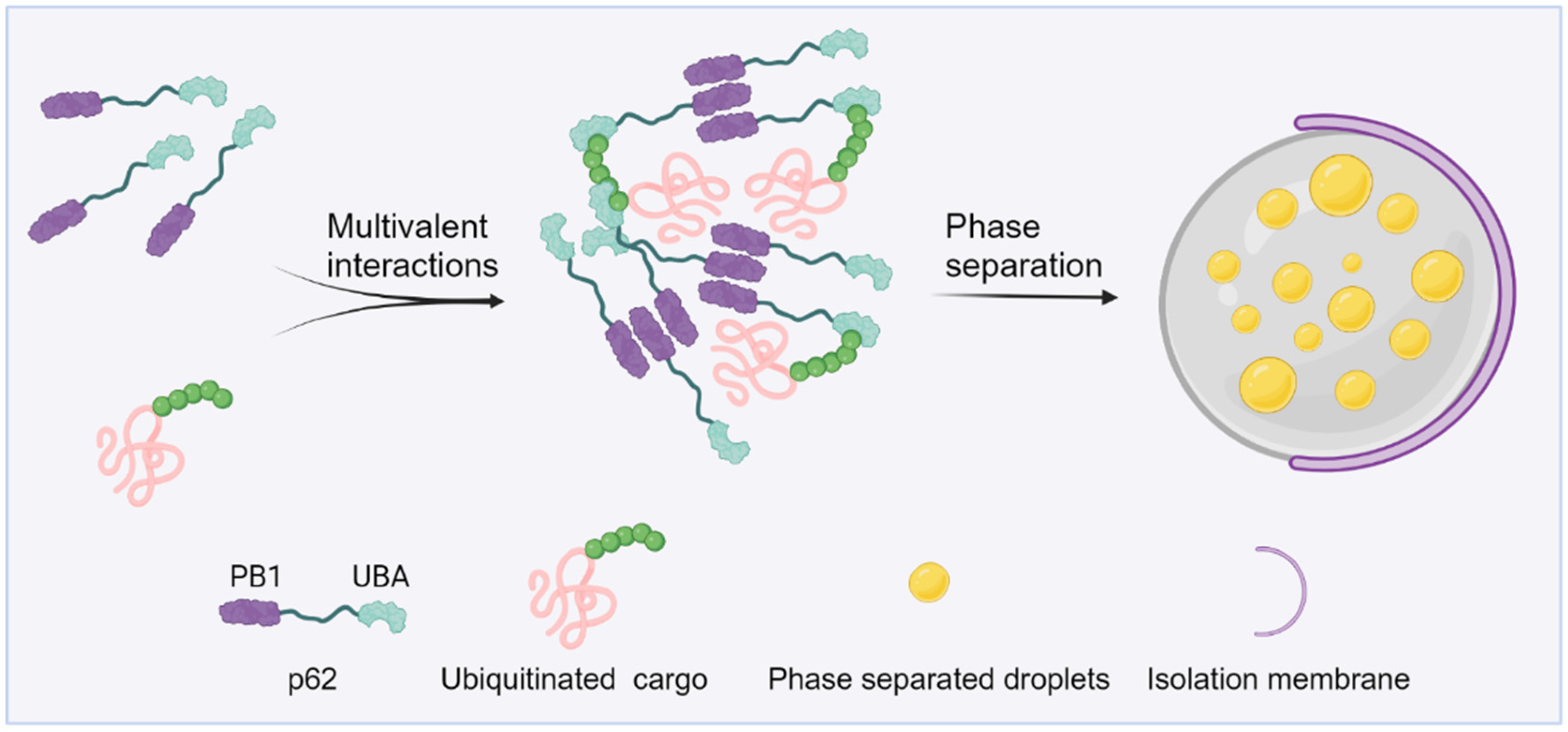
3. Ubiquitin in Autophagy
4. Ubiquitin in Other Biomolecular Condensates
5. Summary and Future Perspectives

{kind=link}
{kind=link}
| Biomolecular Condensates | Ubiquitin | Effect on Phase Separation | Reference |
|---|---|---|---|
| Arsenite, or heat-induced stress granules | Poly-ubiquitin | Promotes LLPS | [30] |
| Heat-induced stress granules | Mono-ubiquitin | Causes disassembly | [28] |
| Arsenite, or heat-induced stress granules | K63 poly-ubiquitin | Causes disassembly | [34,43] |
| p62 condensates | Poly-ubiquitin | Promotes LLPS | [70] |
| p62 condensates | Mono-ubiquitin | Causes disassembly | [69] |
| Proteasome condensate | Mono-ubiquitin | Causes disassembly | [116] |
| UBQLN2 phase separation | Poly-ubiquitin | Causes disassembly | [31] |
| Dvl2 phase separation | Poly-ubiquitin | Promotes LLPS | [104] |
| NEMO phase separation | Poly-ubiquitin | Promotes LLPS | [100] |
Author Contributions
Funding
Conflicts of Interest
References
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Ma, W.; Yang, B.; Lu, H.; Zhou, F.; Zhang, L. Post-translational modifications in liquid-liquid phase separation: A comprehensive review. Mol. Biomed. 2022, 3, 13. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Manohar, S.; Jacob, S.; Wang, J.; Wiechecki, K.A.; Koh, H.W.L.; Simoes, V.; Choi, H.; Vogel, C.; Silva, G.M. Polyubiquitin Chains Linked by Lysine Residue 48 (K48) Selectively Target Oxidized Proteins In Vivo. Antioxid. Redox Signal 2019, 31, 1133–1149. [Google Scholar] [CrossRef]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hubscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S.I. K63 ubiquitination in immune signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. Regulation of NF-kappaB by ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Dosa, A.; Csizmadia, T. The role of K63-linked polyubiquitin in several types of autophagy. Biol. Futur. 2022, 73, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, C.A.; Kashyap, T.R.; Nakasone, M.A.; Krueger, S.; Fushman, D. Unique structural, dynamical, and functional properties of k11-linked polyubiquitin chains. Structure 2013, 21, 1168–1181. [Google Scholar] [CrossRef]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar] [CrossRef] [PubMed]
- Wickliffe, K.E.; Williamson, A.; Meyer, H.J.; Kelly, A.; Rape, M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 2011, 21, 656–663. [Google Scholar] [CrossRef]
- Wu, X.; Lei, C.; Xia, T.; Zhong, X.; Yang, Q.; Shu, H.B. Regulation of TRIF-mediated innate immune response by K27-linked polyubiquitination and deubiquitination. Nat. Commun. 2019, 10, 4115. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Ohn, T.; Anderson, P. The role of posttranslational modifications in the assembly of stress granules. Wiley Interdiscip. Rev. RNA 2010, 1, 486–493. [Google Scholar] [CrossRef]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Bohm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol. Cell 2018, 70, 906–919.e7. [Google Scholar] [CrossRef]
- Xie, X.; Matsumoto, S.; Endo, A.; Fukushima, T.; Kawahara, H.; Saeki, Y.; Komada, M. Deubiquitylases USP5 and USP13 are recruited to and regulate heat-induced stress granules through their deubiquitylating activities. J. Cell Sci. 2018, 131, jcs210856. [Google Scholar] [CrossRef]
- Takahashi, M.; Kitaura, H.; Kakita, A.; Kakihana, T.; Katsuragi, Y.; Onodera, O.; Iwakura, Y.; Nawa, H.; Komatsu, M.; Fujii, M. USP10 Inhibits Aberrant Cytoplasmic Aggregation of TDP-43 by Promoting Stress Granule Clearance. Mol. Cell Biol. 2022, 42, e0039321. [Google Scholar] [CrossRef]
- Kwon, S.; Zhang, Y.; Matthias, P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes. Dev. 2007, 21, 3381–3394. [Google Scholar] [CrossRef]
- Dao, T.P.; Kolaitis, R.M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castaneda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Maxwell, B.A.; Joo, J.H.; Gwon, Y.; Messing, J.; Mishra, A.; Shaw, T.I.; Ward, A.L.; Quan, H.; Sakurada, S.M.; et al. ULK1 and ULK2 Regulate Stress Granule Disassembly Through Phosphorylation and Activation of VCP/p97. Mol. Cell 2019, 74, 742–757.e8. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Sinon, A.N.; Lai, A.; Shi, L.; Lancaster, C.L.; Gokhale, A.; Faundez, V.; Bassell, G.J. Cdh1-APC Regulates Protein Synthesis and Stress Granules in Neurons through an FMRP-Dependent Mechanism. iScience 2020, 23, 101132. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Z.; Kang, Y.; Yi, Q.; Wang, T.; Bai, Y.; Liu, Y. Stress granule homeostasis is modulated by TRIM21-mediated ubiquitination of G3BP1 and autophagy-dependent elimination of stress granules. Autophagy 2023, 19, 1934–1951. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Markmiller, S.; Fulzele, A.; Higgins, R.; Leonard, M.; Yeo, G.W.; Bennett, E.J. Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 2019, 27, 1356–1363.e3. [Google Scholar] [CrossRef]
- Maxwell, B.A.; Gwon, Y.; Mishra, A.; Peng, J.; Nakamura, H.; Zhang, K.; Kim, H.J.; Taylor, J.P. Ubiquitination is essential for recovery of cellular activities after heat shock. Science 2021, 372, eabc3593. [Google Scholar] [CrossRef]
- Tolay, N.; Buchberger, A. Comparative profiling of stress granule clearance reveals differential contributions of the ubiquitin system. Life Sci. Alliance 2021, 4, e202000927. [Google Scholar] [CrossRef]
- Itakura, E.; Zavodszky, E.; Shao, S.; Wohlever, M.L.; Keenan, R.J.; Hegde, R.S. Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol. Cell 2016, 63, 21–33. [Google Scholar] [CrossRef]
- Alexander, E.J.; Ghanbari Niaki, A.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef]
- Peng, G.; Gu, A.; Niu, H.; Chen, L.; Chen, Y.; Zhou, M.; Zhang, Y.; Liu, J.; Cai, L.; Liang, D.; et al. Amyotrophic lateral sclerosis (ALS) linked mutation in Ubiquilin 2 affects stress granule assembly via TIA-1. CNS Neurosci. Ther. 2022, 28, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 13425–13430. [Google Scholar] [CrossRef] [PubMed]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 2021, 372, eabf6548. [Google Scholar] [CrossRef] [PubMed]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Muller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef] [PubMed]
- Ripin, N.; Parker, R. Are stress granules the RNA analogs of misfolded protein aggregates? RNA 2022, 28, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, W.K. Tau liquid-liquid phase separation in neurodegenerative diseases. Trends Cell Biol. 2022, 32, 611–623. [Google Scholar] [CrossRef]
- Parolini, F.; Tira, R.; Barracchia, C.G.; Munari, F.; Capaldi, S.; D’Onofrio, M.; Assfalg, M. Ubiquitination of Alzheimer’s-related tau protein affects liquid-liquid phase separation in a site- and cofactor-dependent manner. Int. J. Biol. Macromol. 2022, 201, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E.A.; Kraus, F.; Harper, J.W. Mechanisms underlying ubiquitin-driven selective mitochondrial and bacterial autophagy. Mol. Cell 2022, 82, 1501–1513. [Google Scholar] [CrossRef]
- Chu, Y.; Kang, Y.; Yan, C.; Yang, C.; Zhang, T.; Huo, H.; Liu, Y. LUBAC and OTULIN regulate autophagy initiation and maturation by mediating the linear ubiquitination and the stabilization of ATG13. Autophagy 2021, 17, 1684–1699. [Google Scholar] [CrossRef]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol. Cell 2016, 61, 84–97. [Google Scholar] [CrossRef]
- Fujioka, Y.; Noda, N.N. Biomolecular condensates in autophagy regulation. Curr. Opin. Cell Biol. 2021, 69, 23–29. [Google Scholar] [CrossRef]
- Noda, N.N.; Wang, Z.; Zhang, H. Liquid-liquid phase separation in autophagy. J. Cell Biol. 2020, 219, e202004062. [Google Scholar] [CrossRef]
- Lu, Y.; Chang, C. Phase Separation in Regulation of Autophagy. Front. Cell Dev. Biol. 2022, 10, 910640. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.; Alam, J.M.; Noshiro, D.; Hirata, E.; Fujioka, Y.; Suzuki, K.; Ohsumi, Y.; Noda, N.N. Liquidity Is a Critical Determinant for Selective Autophagy of Protein Condensates. Mol. Cell 2020, 77, 1163–1175.e9. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Alam, J.M.; Noshiro, D.; Mouri, K.; Ando, T.; Okada, Y.; May, A.I.; Knorr, R.L.; Suzuki, K.; Ohsumi, Y.; et al. Phase separation organizes the site of autophagosome formation. Nature 2020, 578, 301–305. [Google Scholar] [CrossRef]
- Shi, X.; Chang, C.; Yokom, A.L.; Jensen, L.E.; Hurley, J.H. The autophagy adaptor NDP52 and the FIP200 coiled-coil allosterically activate ULK1 complex membrane recruitment. Elife 2020, 9, e59099. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Danieli, A.; Martens, S. p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J. Cell Sci. 2018, 131, jcs214304. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Savova, A.; Danieli, A.; Romanov, J.; Tremel, S.; Ebner, M.; Peterbauer, T.; Sztacho, M.; Trapannone, R.; Tarafder, A.K.; et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018, 37, e98308. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.; Johansen, T.; Sachse, C. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Dikic, I. Ubiquitin-induced phase separation of p62/SQSTM1. Cell Res. 2018, 28, 389–390. [Google Scholar] [CrossRef]
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017, 27, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, N.L.; Kournoutis, A.; Lamark, T.; Johansen, T. NBR1: The archetypal selective autophagy receptor. J. Cell Biol. 2022, 221, e202208092. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Ferrari, L.; Martens, S. Orchestration of selective autophagy by cargo receptors. Curr. Biol. 2022, 32, R1357–R1371. [Google Scholar] [CrossRef]
- Turco, E.; Savova, A.; Gere, F.; Ferrari, L.; Romanov, J.; Schuschnig, M.; Martens, S. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat. Commun. 2021, 12, 5212. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Peng, S.Z.; Chen, X.H.; Chen, S.J.; Zhang, J.; Wang, C.Y.; Liu, W.R.; Zhang, D.; Su, Y.; Zhang, X.K. Phase separation of Nur77 mediates celastrol-induced mitophagy by promoting the liquidity of p62/SQSTM1 condensates. Nat. Commun. 2021, 12, 5989. [Google Scholar] [CrossRef]
- Odeh, H.M.; Shorter, J. Aggregates of TDP-43 protein spiral into view. Nature 2022, 601, 29–30. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillee, S.; Doukoure, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.; Morris, A.; de Belleroche, J.S. Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: Prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur. J. Hum. Genet. 2014, 22, 492–496. [Google Scholar] [CrossRef]
- Dessay, M.; Jobin Gervais, F.; Simonyan, D.; Samson, A.; Gleeton, G.; Gagnon, E.; Albert, C.; Brown, J.P.; Michou, L. Clinical phenotype of adult offspring carriers of the p.Pro392Leu mutation within the SQSTM1 gene in Paget’s disease of bone. Bone Rep. 2020, 13, 100717. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Walsh, J.P.; Layfield, R.; Ratajczak, T.; Xu, J. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr. Rev. 2013, 34, 501–524. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef]
- Spector, D.L.; Lamond, A.I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 2011, 3, a000646. [Google Scholar] [CrossRef]
- Ilik, I.A.; Malszycki, M.; Lubke, A.K.; Schade, C.; Meierhofer, D.; Aktas, T. SON and SRRM2 are essential for nuclear speckle formation. Elife 2020, 9, e60579. [Google Scholar] [CrossRef]
- Xu, S.; Lai, S.K.; Sim, D.Y.; Ang, W.S.L.; Li, H.Y.; Roca, X. SRRM2 organizes splicing condensates to regulate alternative splicing. Nucleic Acids Res. 2022, 50, 8599–8614. [Google Scholar] [CrossRef]
- Cuneo, M.J.; Mittag, T. The ubiquitin ligase adaptor SPOP in cancer. FEBS J. 2019, 286, 3946–3958. [Google Scholar] [CrossRef]
- Bouchard, J.J.; Otero, J.H.; Scott, D.C.; Szulc, E.; Martin, E.W.; Sabri, N.; Granata, D.; Marzahn, M.R.; Lindorff-Larsen, K.; Salvatella, X.; et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 2018, 72, 19–36.e8. [Google Scholar] [CrossRef] [PubMed]
- Marzahn, M.R.; Marada, S.; Lee, J.; Nourse, A.; Kenrick, S.; Zhao, H.; Ben-Nissan, G.; Kolaitis, R.M.; Peters, J.L.; Pounds, S.; et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016, 35, 1254–1275. [Google Scholar] [CrossRef]
- Liu, S.; Wang, T.; Shi, Y.; Bai, L.; Wang, S.; Guo, D.; Zhang, Y.; Qi, Y.; Chen, C.; Zhang, J.; et al. USP42 drives nuclear speckle mRNA splicing via directing dynamic phase separation to promote tumorigenesis. Cell Death Differ. 2021, 28, 2482–2498. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Laporte, D.; Salin, B.; Daignan-Fornier, B.; Sagot, I. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J. Cell Biol. 2008, 181, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.C.; Wu, E.; Sailer, C.; Jando, J.; Styles, E.; Eisenkolb, I.; Kuschel, M.; Bitschar, K.; Wang, X.; Huang, L.; et al. Ubiquitin orchestrates proteasome dynamics between proliferation and quiescence in yeast. Mol. Biol. Cell 2017, 28, 2479–2491. [Google Scholar] [CrossRef]
- Laporte, D.; Lebaudy, A.; Sahin, A.; Pinson, B.; Ceschin, J.; Daignan-Fornier, B.; Sagot, I. Metabolic status rather than cell cycle signals control quiescence entry and exit. J. Cell Biol. 2011, 192, 949–957. [Google Scholar] [CrossRef]
- Enenkel, C.; Kang, R.W.; Wilfling, F.; Ernst, O.P. Intracellular localization of the proteasome in response to stress conditions. J. Biol. Chem. 2022, 298, 102083. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Du, M.; Ea, C.K.; Fang, Y.; Chen, Z.J. Liquid phase separation of NEMO induced by polyubiquitin chains activates NF-kappaB. Mol. Cell 2022, 82, 2415–2426.e5. [Google Scholar] [CrossRef]
- Goel, S.; Oliva, R.; Jeganathan, S.; Bader, V.; Krause, L.J.; Kriegler, S.; Stender, I.D.; Christine, C.W.; Nakamura, K.; Hoffmann, J.E.; et al. Linear ubiquitination induces NEMO phase separation to activate NF-kappaB signaling. Life Sci. Alliance 2023, 6, e202201607. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Hao, W.; Wang, L.; Li, S. Linear Ubiquitination Mediates EGFR-Induced NF-kappaB Pathway and Tumor Development. Int. J. Mol. Sci. 2021, 22, 11875. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Gao, Y.; Wang, H.Y. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal 2008, 20, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Vamadevan, V.; Chaudhary, N.; Maddika, S. Ubiquitin-assisted phase separation of dishevelled-2 promotes Wnt signalling. J. Cell Sci. 2022, 135, jcs260284. [Google Scholar] [CrossRef]
- LoPresti, P. HDAC6 in Diseases of Cognition and of Neurons. Cells 2020, 10, 12. [Google Scholar] [CrossRef]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef]
- Trzeciakiewicz, H.; Ajit, D.; Tseng, J.H.; Chen, Y.; Ajit, A.; Tabassum, Z.; Lobrovich, R.; Peterson, C.; Riddick, N.V.; Itano, M.S.; et al. An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun. 2020, 11, 5522. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Yoo, Y.E.; Ko, C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef]
- Ma, S.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.M.; Chung, R.S.; Lee, A. The converging roles of sequestosome-1/p62 in the molecular pathways of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Neurobiol. Dis. 2022, 166, 105653. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef]
- Bello, A.I.; Goswami, R.; Brown, S.L.; Costanzo, K.; Shores, T.; Allan, S.; Odah, R.; Mohan, R.D. Deubiquitinases in Neurodegeneration. Cells 2022, 11, 556. [Google Scholar] [CrossRef]
- Song, A.; Hazlett, Z.; Abeykoon, D.; Dortch, J.; Dillon, A.; Curtiss, J.; Martinez, S.B.; Hill, C.P.; Yu, C.; Huang, L.; et al. Branched ubiquitin chain binding and deubiquitination by UCH37 facilitate proteasome clearance of stress-induced inclusions. eLife 2021, 10, e72798. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, P.; Zhang, J.; Wang, B. Emerging Roles of Ubiquitination in Biomolecular Condensates. Cells 2023, 12, 2329. https://doi.org/10.3390/cells12182329
Liang P, Zhang J, Wang B. Emerging Roles of Ubiquitination in Biomolecular Condensates. Cells. 2023; 12(18):2329. https://doi.org/10.3390/cells12182329
Chicago/Turabian StyleLiang, Peigang, Jiaqi Zhang, and Bo Wang. 2023. "Emerging Roles of Ubiquitination in Biomolecular Condensates" Cells 12, no. 18: 2329. https://doi.org/10.3390/cells12182329