
Astrocytes in Parkinson’s Disease: From Role to Possible Intervention

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Beneficial Roles of Astrocytes to Neuronal Survival
2.1. Astrocytes Secrete Various Molecules That Are Beneficial to Neuronal Survival
2.2. Removal of α-Synuclein
2.3. Healthy Astrocyte Mitochondria Are Beneficial to Cellular Survival
2.3.1. The Transfer of Healthy Mitochondria between Neurons and Astrocytes Is Beneficial
2.3.2. Metabolism of Fatty Acids
2.4. Excitotoxicity and Glutamate Metabolism
2.5. Role in the Blood–Brain Barrier

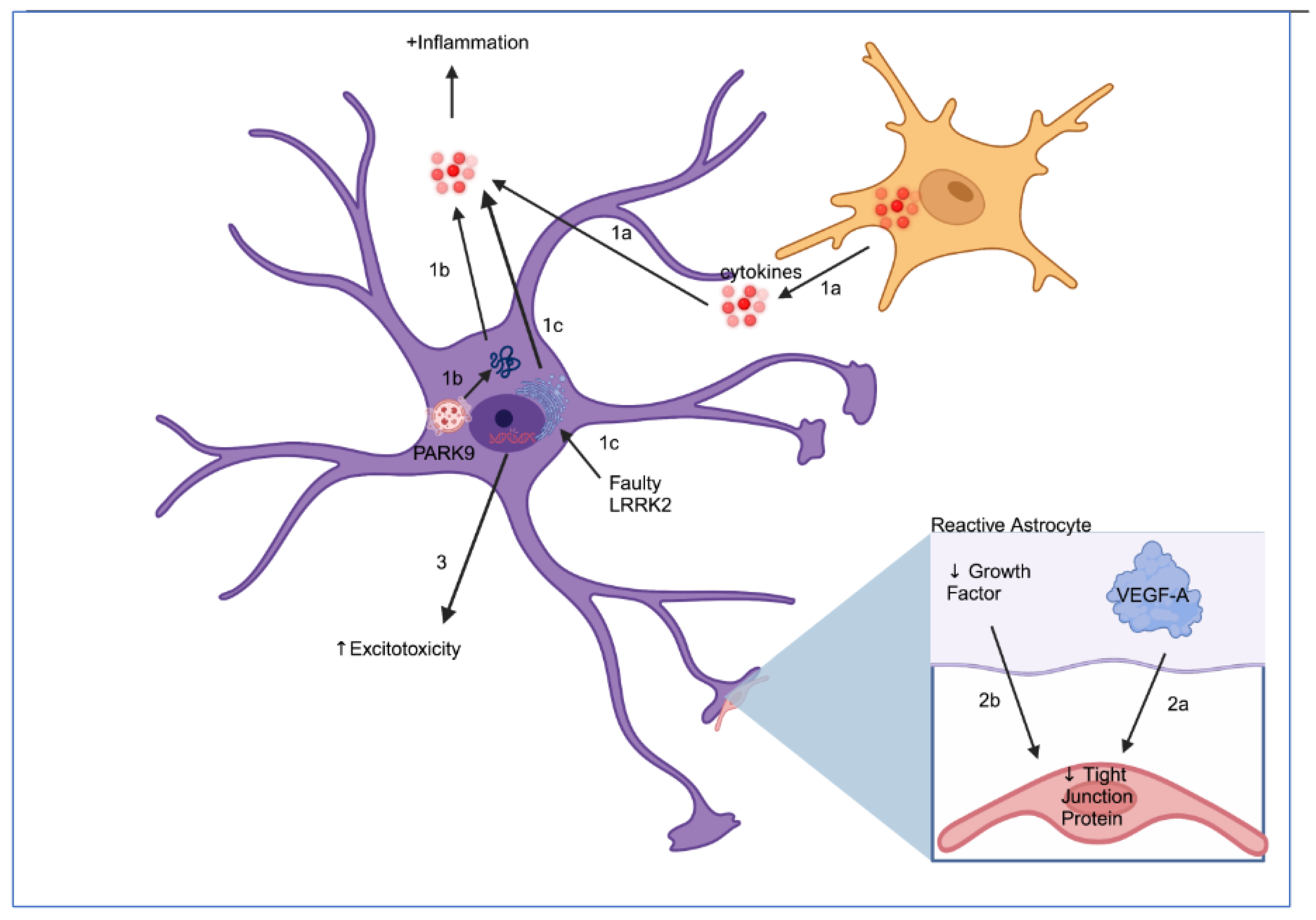
3. Neurotoxic Activities of Astrocytes
3.1. Glutamate Metabolism
3.2. Secretion of Inflammatory Cytokines
3.3. Blood–Brain Barrier Disruption
4. Astrocyte Therapy in PD
4.1. Promoting the Secretion of Neurotrophic Factors
4.2. Prevention of Astrocyte Conversion into a Pro-Inflammatory Phenotype and Inflammatory Response
4.3. Astrocyte Graft
4.4. Astrocyte Reprogramming
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, E.R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2022 Update. J. Park. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of Autophagy and Mitochondrial Function in Parkinson’s Disease. Transl. Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.-S. Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers with a Common Mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Yang, T.; Gu, Y.; Sun, X.-H. Mitochondrial Dysfunction in Parkinson’s Disease: From Mechanistic Insights to Therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal Numbers of Neuronal and Nonneuronal Cells Make the Human Brain an Isometrically Scaled-up Primate Brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Petrova, P.S.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. Discovering Novel Phenotype-Selective Neurotrophic Factors to Treat Neurodegenerative Diseases. Prog. Brain Res. 2004, 146, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Kostuk, E.W.; Cai, J.; Iacovitti, L. Subregional Differences in Astrocytes Underlie Selective Neurodegeneration or Protection in Parkinson’s Disease Models in Culture. Glia 2019, 67, 1542–1557. [Google Scholar] [CrossRef]
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The Role of Astrocytes in the Neurorepair Process. Front. Cell Dev. Biol. 2021, 9, 665795. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Zhou, K.-G.; Yin, Y.-X.; Jin, L.-J.; Tong, W.-F.; Guo, J.; Yu, L.-H.; Ye, X.-C.; Jiang, M. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Prevents the Neuroinflammation Induced Dopaminergic Neurodegeneration. Exp. Gerontol. 2023, 171, 112037. [Google Scholar] [CrossRef]
- Deierborg, T.; Soulet, D.; Roybon, L.; Hall, V.; Brundin, P. Emerging Restorative Treatments for Parkinson’s Disease. Prog. Neurobiol. 2008, 85, 407–432. [Google Scholar] [CrossRef]
- Lin, L.-F.H.; Zhang, T.J.; Collins, F.; Armes, L.G. Purification and Initial Characterization of Rat B49 Glial Cell Line-Derived Neurotrophic Factor. J. Neurochem. 1994, 63, 758–768. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF Family: Signalling, Biological Functions and Therapeutic Value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Grothe, C.; Timmer, M. The Physiological and Pharmacological Role of Basic Fibroblast Growth Factor in the Dopaminergic Nigrostriatal System. Brain Res. Rev. 2007, 54, 80–91. [Google Scholar] [CrossRef]
- Richter, C. Reactive Oxygen and DNA Damage in Mitochondria. Mutat. Res. 1992, 275, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hirrlinger, J. Glutathione Pathways in the Brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Schuele, L.-L.; Schuermann, B.; Bilkei-Gorzo, A.; Gorgzadeh, S.; Zimmer, A.; Leidmaa, E. Regulation of Adult Neurogenesis by the Endocannabinoid-Producing Enzyme Diacylglycerol Lipase Alpha (DAGLa). Sci. Rep. 2022, 12, 633. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.; Chen, C. Endocannabinoid 2-Arachidonoylglycerol Protects Neurons against β-Amyloid Insults. Neuroscience 2011, 178, 159–168. [Google Scholar] [CrossRef]
- Dettmer, U. Rationally Designed Variants of α-Synuclein Illuminate Its In Vivo Structural Properties in Health and Disease. Front. Neurosci. 2018, 12, 623. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Roberts, H.L.; Brown, D.R. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef]
- Da Fonseca, T.L.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef]
- Jang, A.; Lee, H.-J.; Suk, J.-E.; Jung, J.-W.; Kim, K.-P.; Lee, S.-J. Non-Classical Exocytosis of Alpha-Synuclein Is Sensitive to Folding States and Promoted under Stress Conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef]
- Xie, Y.X.; Naseri, N.N.; Fels, J.; Kharel, P.; Na, Y.; Lane, D.; Burré, J.; Sharma, M. Lysosomal Exocytosis Releases Pathogenic α-Synuclein Species from Neurons in Synucleinopathy Models. Nat. Commun. 2022, 13, 4918. [Google Scholar] [CrossRef]
- Yang, Y.; Song, J.-J.; Choi, Y.R.; Kim, S.-H.; Seok, M.-J.; Wulansari, N.; Darsono, W.H.W.; Kwon, O.-C.; Chang, M.-Y.; Park, S.M.; et al. Therapeutic Functions of Astrocytes to Treat α-Synuclein Pathology in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2110746119. [Google Scholar] [CrossRef]
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the Astrocytic Clearance of Extracellular α-Synuclein Aggregates by Ginkgolides Attenuates Neural Cell Injury. Cell. Mol. Neurobiol. 2019, 39, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Qiao, X.; Matos, J.O.; Farley, L.; Cilenti, L.; Chen, B.; Tatulian, S.A.; Teter, K. Reversal of Alpha-Synuclein Fibrillization by Protein Disulfide Isomerase. Front. Cell Dev. Biol. 2020, 8, 726. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.-L.; Atkins, C.; Daniels, B.P. Fibrillar α-Synuclein Induces Neurotoxic Astrocyte Activation via RIP Kinase Signaling and NF-ΚB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive Uptake of α-Synuclein Oligomers in Astrocytes Results in Sustained Intracellular Deposits and Mitochondrial Damage. Mol. Cell Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Bruzzone, S.; Verderio, C.; Schenk, U.; Fedele, E.; Zocchi, E.; Matteoli, M.; De Flora, A. Glutamate-Mediated Overexpression of CD38 in Astrocytes Cultured with Neurones. J. Neurochem. 2004, 89, 264–272. [Google Scholar] [CrossRef] [PubMed]
- English, K.; Shepherd, A.; Uzor, N.-E.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes Rescue Neuronal Health after Cisplatin Treatment through Mitochondrial Transfer. Acta Neuropathol. Commun. 2020, 8, 36. [Google Scholar] [CrossRef]
- Cheng, X.-Y.; Biswas, S.; Li, J.; Mao, C.-J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.-R.; Liu, C.-F.; et al. Human IPSCs Derived Astrocytes Rescue Rotenone-Induced Mitochondrial Dysfunction and Dopaminergic Neurodegeneration in Vitro by Donating Functional Mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Ralhan, I.; Chang, C.-L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid Droplets in the Nervous System. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Why Does Brain Metabolism Not Favor Burning of Fatty Acids to Provide Energy?—Reflections on Disadvantages of the Use of Free Fatty Acids as Fuel for Brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid Homeostasis, Lipotoxicity and the Metabolic Syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214.e5. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Magistretti, P.J. The Role of Astroglia in Neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [CrossRef]
- Hofmann, K.; Rodriguez-Rodriguez, R.; Gaebler, A.; Casals, N.; Scheller, A.; Kuerschner, L. Astrocytes and Oligodendrocytes in Grey and White Matter Regions of the Brain Metabolize Fatty Acids. Sci. Rep. 2017, 7, 10779. [Google Scholar] [CrossRef]
- Dzamba, D.; Honsa, P.; Anderova, M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013, 11, 250–262. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Ye, C.; Ge, W.; Chen, Y.; Jiang, Z.; Wu, C.; Poo, M.; Duan, S. ATP Released by Astrocytes Mediates Glutamatergic Activity-Dependent Heterosynaptic Suppression. Neuron 2003, 40, 971–982. [Google Scholar] [CrossRef]
- Koizumi, S.; Fujishita, K.; Tsuda, M.; Shigemoto-Mogami, Y.; Inoue, K. Dynamic Inhibition of Excitatory Synaptic Transmission by Astrocyte-Derived ATP in Hippocampal Cultures. Proc. Natl. Acad. Sci. USA 2003, 100, 11023–11028. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.N.; Pankratov, Y. ATP from Synaptic Terminals and Astrocytes Regulates NMDA Receptors and Synaptic Plasticity through PSD-95 Multi-Protein Complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate Excitotoxicity: Potential Therapeutic Target for Ischemic Stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Eulenburg, V.; Gomeza, J. Neurotransmitter Transporters Expressed in Glial Cells as Regulators of Synapse Function. Brain Res. Rev. 2010, 63, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Lehre, K.P.; Danbolt, N.C. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.A.; Aizenman, E. Hundred-Fold Increase in Neuronal Vulnerability to Glutamate Toxicity in Astrocyte-Poor Cultures of Rat Cerebral Cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Cho, Y.; Bannai, S. Uptake of Glutamate and Cystine in C-6 Glioma Cells and in Cultured Astrocytes. J. Neurochem. 1990, 55, 2091–2097. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte Glutamate Transport: Review of Properties, Regulation, and Physiological Functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and Exacerbation of Brain Injury in Mice Lacking the Glutamate Transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 Glutamate Transporters. Pflug. Arch. Eur. J. Physiol. 2014, 466, 3–24. [Google Scholar] [CrossRef]
- McKenna, M.C.; Stridh, M.H.; McNair, L.F.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Glutamate Oxidation in Astrocytes: Roles of Glutamate Dehydrogenase and Aminotransferases. J. Neurosci. Res. 2016, 94, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Waniewski, R.A.; Martin, D.L. Exogenous Glutamate Is Metabolized to Glutamine and Exported by Rat Primary Astrocyte Cultures. J. Neurochem. 1986, 47, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef]
- Aoki, C.; Milner, T.A.; Berger, S.B.; Sheu, K.F.; Blass, J.P.; Pickel, V.M. Glial Glutamate Dehydrogenase: Ultrastructural Localization and Regional Distribution in Relation to the Mitochondrial Enzyme, Cytochrome Oxidase. J. Neurosci. Res. 1987, 18, 305–318. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Back, A.; Tupper, K.Y.; Bai, T.; Chiranand, P.; Goldenberg, F.D.; Frank, J.I.; Brorson, J.R. Ammonia-Induced Brain Swelling and Neurotoxicity in an Organotypic Slice Model. Neurol. Res. 2011, 33, 1100–1108. [Google Scholar] [CrossRef]
- Bröer, S.; Brookes, N. Transfer of Glutamine between Astrocytes and Neurons. J. Neurochem. 2001, 77, 705–719. [Google Scholar] [CrossRef]
- Hertz, L.; Dringen, R.; Schousboe, A.; Robinson, S.R. Astrocytes: Glutamate Producers for Neurons. J. Neurosci. Res. 1999, 57, 417–428. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The Glutamate/GABA-Glutamine Cycle: Aspects of Transport, Neurotransmitter Homeostasis and Ammonia Transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Schousboe, A.; Sarup, A.; Bak, L.K.; Waagepetersen, H.S.; Larsson, O.M. Role of Astrocytic Transport Processes in Glutamatergic and GABAergic Neurotransmission. Neurochem. Int. 2004, 45, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nomura, M.; Yamagishi, S.; Harada, S.; Yamashita, J.; Yamamoto, H. Induction of Various Blood-Brain Barrier Properties in Non-Neural Endothelial Cells by Close Apposition to Co-Cultured Astrocytes. Glia 1997, 19, 13–26. [Google Scholar] [CrossRef]
- Pivoriūnas, A.; Verkhratsky, A. Astrocyte–Endotheliocyte Axis in the Regulation of the Blood–Brain Barrier. Neurochem. Res. 2021, 46, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.L.; Leach, L.; Clarke, G.J.; Nolan, C.C.; Ray, D.E. Reversible Disruption of Tight Junction Complexes in the Rat Blood-Brain Barrier, Following Transitory Focal Astrocyte Loss. Glia 2004, 48, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Lippoldt, A. Tight Junctions of the Blood–Brain Barrier: Development, Composition and Regulation. Vasc. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.V. Tight Junction Protein Expression in Human Astrocytes. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2016. Available online: https://etheses.whiterose.ac.uk/14403/ (accessed on 18 July 2023).
- Lanciotti, A.; Brignone, M.; Bertini, E.; Petrucci, T.; Aloisi, F.; Ambrosini, E. Astrocytes: Emerging Stars in Leukodystrophy Pathogenesis. Transl. Neurosci. 2013, 4, 144–164. [Google Scholar] [CrossRef]
- Lisjak, M.; Potokar, M.; Rituper, B.; Jorgačevski, J.; Zorec, R. AQP4e-Based Orthogonal Arrays Regulate Rapid Cell Volume Changes in Astrocytes. J. Neurosci. 2017, 37, 10748–10756. [Google Scholar] [CrossRef]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased Seizure Duration and Slowed Potassium Kinetics in Mice Lacking Aquaporin-4 Water Channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef]
- Agarwal, R.; Shukla, G.S. Potential Role of Cerebral Glutathione in the Maintenance of Blood-Brain Barrier Integrity in Rat. Neurochem. Res. 1999, 24, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Giusti, V.; Pischedda, F.; Giusto, E.; Plotegher, N.; Marte, A.; Battisti, I.; Di Iacovo, A.; Marku, A.; Piccoli, G.; et al. Trafficking of the Glutamate Transporter Is Impaired in LRRK2-Related Parkinson’s Disease. Acta Neuropathol. 2022, 144, 81–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Cha, S.-H.; Choi, Y.R.; Jou, I.; Joe, E.-H.; Park, S.M. DJ-1 Deficiency Impairs Glutamate Uptake into Astrocytes via the Regulation of Flotillin-1 and Caveolin-1 Expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.S.; Park, J.-Y.; Suh, Y.H.; Jou, I.; Joe, E.-H.; Park, S.M. DJ-1 Associates with Lipid Rafts by Palmitoylation and Regulates Lipid Rafts-Dependent Endocytosis in Astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R.; Tian, G.; Guo, H.; Lin, C.G. Association of Excitatory Amino Acid Transporters, Especially EAAT2, with Cholesterol-Rich Lipid Raft Microdomains: Importance for Excitatory Amino Acid Transporter Localization and Function. J. Biol. Chem. 2004, 279, 34388–34396. [Google Scholar] [CrossRef] [PubMed]
- Ferrarese, C.; Zoia, C.; Pecora, N.; Piolti, R.; Frigo, M.; Bianchi, G.; Sala, G.; Begni, B.; Riva, R.; Frattola, L. Reduced Platelet Glutamate Uptake in Parkinson’s Disease. J. Neural Transm. 1999, 106, 685–692. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and Transforming Growth Factor-α Levels Are Elevated in Ventricular Cerebrospinal Fluid in Juvenile Parkinsonism and Parkinson’s Disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor Necrosis Factor-α (TNF-α) Increases Both in the Brain and in the Cerebrospinal Fluid from Parkinsonian Patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming Growth Factor-Β1 Levels Are Elevated in the Striatum and in Ventricular Cerebrospinal Fluid in Parkinson’s Disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Aloe, L.; Fiore, M. TNF-α Expressed in the Brain of Transgenic Mice Lowers Central Tyroxine Hydroxylase Immunoreactivity and Alters Grooming Behavior. Neurosci. Lett. 1997, 238, 65–68. [Google Scholar] [CrossRef]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine Hydroxylase and Regulation of Dopamine Synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct Transfer of Alpha-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Y. Mutations in the ATP13A2 Gene and Parkinsonism: A Preliminary Review. Biomed. Res. Int. 2014, 2014, 371256. [Google Scholar] [CrossRef]
- Fujii, T.; Nagamori, S.; Wiriyasermkul, P.; Zheng, S.; Yago, A.; Shimizu, T.; Tabuchi, Y.; Okumura, T.; Fujii, T.; Takeshima, H.; et al. Parkinson’s Disease-Associated ATP13A2/PARK9 Functions as a Lysosomal H+,K+-ATPase. Nat. Commun. 2023, 14, 2174. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, J.-H.; Kim, H.; Park, S.M.; Joe, E.-H.; Jou, I. Parkinson’s Disease-Associated LRRK2-G2019S Mutant Acts through Regulation of SERCA Activity to Control ER Stress in Astrocytes. Acta Neuropathol. Commun. 2019, 7, 68. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A Modulator of Wild-Type Glucocerebrosidase Improves Pathogenic Phenotypes in Dopaminergic Neuronal Models of Parkinson’s Disease. Sci. Transl. Med. 2019, 11, eaau6870. [Google Scholar] [CrossRef]
- Mahoney-Crane, C.L.; Viswanathan, M.; Russell, D.; Curtiss, R.A.C.; Freire, J.; Bobba, S.S.; Coyle, S.D.; Kandebo, M.; Yao, L.; Wan, B.-L.; et al. Neuronopathic GBA1L444P Mutation Accelerates Glucosylsphingosine Levels and Formation of Hippocampal Alpha-Synuclein Inclusions. J. Neurosci. 2023, 43, 501–521. [Google Scholar] [CrossRef]
- Galvagnion, C.; Marlet, F.R.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid Changes in Parkinson L444P GBA Mutation Fibroblasts Promote α-Synuclein Aggregation. Brain 2022, 145, 1038–1051. [Google Scholar] [CrossRef]
- Sanyal, A.; DeAndrade, M.P.; Novis, H.S.; Lin, S.; Chang, J.; Lengacher, N.; Tomlinson, J.J.; Tansey, M.G.; LaVoie, M.J. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov. Disord. 2020, 35, 760–773. [Google Scholar] [CrossRef]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.-H.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of Astroglial Nuclear Factor ΚB Reduces Inflammation and Improves Functional Recovery after Spinal Cord Injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear Factor-Kappa β as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garcia-Garrote, M.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. NRF2 Activation and Downstream Effects: Focus on Parkinson’s Disease and Brain Angiotensin. Antioxidants 2021, 10, 1649. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, R.; Zhang, F. Role of Nrf2 in Parkinson’s Disease: Toward New Perspectives. Front. Pharmacol. 2022, 13, 919233. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.-K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-ΚB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2022, 9, 809952. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ong, W.-Y.; Horrocks, L.A. Inhibitors of Brain Phospholipase A2 Activity: Their Neuropharmacological Effects and Therapeutic Importance for the Treatment of Neurologic Disorders. Pharmacol. Rev. 2006, 58, 591–620. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.-Y. NLR Members NLRC4 and NLRP3 Mediate Sterile Inflammasome Activation in Microglia and Astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Barclay, W.E.; Aggarwal, N.; Deerhake, M.E.; Inoue, M.; Nonaka, T.; Nozaki, K.; Luzum, N.A.; Miao, E.A.; Shinohara, M.L. The AIM2 Inflammasome Is Activated in Astrocytes during the Late Phase of EAE. JCI Insight 2022, 7, e155563. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood–Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes Are Necessary for Blood-Brain Barrier Maintenance in the Adult Mouse Brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Cassina, P.; Vargas, M.R.; Castellanos, R.; Viera, L.; Beckman, J.S.; Estévez, A.G.; Barbeito, L. Astrocytic Production of Nerve Growth Factor in Motor Neuron Apoptosis: Implications for Amyotrophic Lateral Sclerosis. J. Neurochem. 2004, 89, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-Mediated Disruption of Endothelial CLN-5 Promotes Blood-Brain Barrier Breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-Brain Barrier Dysfunction in Parkinsonian Midbrain in Vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R.; Lozano, A.M.; Penn, R.D.; Simpson, R.K.; Stacy, M.; et al. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor. Randomized, Double-Blind Trial of Glial Cell Line-Derived Neurotrophic Factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized Controlled Trial of Intraputamenal Glial Cell Line–Derived Neurotrophic Factor Infusion in Parkinson Disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Bäck, S.; Peränen, J.; Galli, E.; Pulkkila, P.; Lonka-Nevalaita, L.; Tamminen, T.; Voutilainen, M.H.; Raasmaja, A.; Saarma, M.; Männistö, P.T.; et al. Gene Therapy with AAV2-CDNF Provides Functional Benefits in a Rat Model of Parkinson’s Disease. Brain Behav. 2013, 3, 75–88. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Lin, Q.; Collier, T.J.; Frim, D.M.; Breakefield, X.O.; Bohn, M.C. Astrocytes Retrovirally Transduced with BDNF Elicit Behavioral Improvement in a Rat Model of Parkinson’s Disease. Brain Res. 1995, 691, 25–36. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene Delivery of AAV2-Neurturin for Parkinson’s Disease: A Double-Blind, Randomised, Controlled Trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Cordero-Llana, Ó.; Houghton, B.C.; Rinaldi, F.; Taylor, H.; Yáñez-Muñoz, R.J.; Uney, J.B.; Wong, L.-F.; Caldwell, M.A. Enhanced Efficacy of the CDNF/MANF Family by Combined Intranigral Overexpression in the 6-OHDA Rat Model of Parkinson’s Disease. Mol. Ther. 2015, 23, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Wang, Y.; He, Y.; Wang, T.; Huang, X.-H.; Zhao, C.-M.; Zhang, L.; Li, S.-W.; Wang, C.; Qu, Y.-N.; et al. A1 Astrocytes Contribute to Murine Depression-like Behavior and Cognitive Dysfunction, Which Can Be Alleviated by IL-10 or Fluorocitrate Treatment. J. Neuroinflamm. 2020, 17, 200. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Baek, J.Y.; Kim, S.R.; Ko, H.W.; Bok, E.; Shin, W.-H.; Won, S.-Y.; Jin, B.K. Capsaicin Prevents Degeneration of Dopamine Neurons by Inhibiting Glial Activation and Oxidative Stress in the MPTP Model of Parkinson’s Disease. Exp. Mol. Med. 2017, 49, e298. [Google Scholar] [CrossRef]
- Du, R.-W.; Bu, W.-G. Simvastatin Prevents Neurodegeneration in the MPTP Mouse Model of Parkinson’s Disease via Inhibition of A1 Reactive Astrocytes. Neuroimmunomodulation 2021, 28, 82–89. [Google Scholar] [CrossRef]
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin Inhibits Activation of NADPH Oxidase/P38 MAPK Pathway and Enhances Expression of Antioxidant Protein in Parkinson Disease Models. Front. Mol. Neurosci. 2018, 11, 165. [Google Scholar] [CrossRef]
- Stevens, K.N.; Creanor, S.; Jeffery, A.; Whone, A.; Zajicek, J.; Foggo, A.; Jones, B.; Chapman, R.; Cocking, L.; Wilks, J.; et al. Evaluation of Simvastatin as a Disease-Modifying Treatment for Patients with Parkinson Disease. JAMA Neurol. 2022, 79, 1232–1241. [Google Scholar] [CrossRef]
- Liu, G.; Sterling, N.W.; Kong, L.; Lewis, M.M.; Mailman, R.B.; Chen, H.; Leslie, D.; Huang, X. Statins May Facilitate Parkinson’s Disease: Insight Gained from a Large, National Claims Database. Mov. Disord. 2017, 32, 913–917. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of Astrocytic Form and Function. In Astrocytes: Methods and Protocols; Milner, R., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 23–45. [Google Scholar] [CrossRef]
- Proschel, C.; Stripay, J.L.; Shih, C.-H.; Munger, J.C.; Noble, M.D. Delayed Transplantation of Precursor Cell-Derived Astrocytes Provides Multiple Benefits in a Rat Model of Parkinsons. EMBO Mol. Med. 2014, 6, 504–518. [Google Scholar] [CrossRef]
- Bahat-Stroomza, M.; Barhum, Y.; Levy, Y.S.; Karpov, O.; Bulvik, S.; Melamed, E.; Offen, D. Induction of Adult Human Bone Marrow Mesenchymal Stromal Cells into Functional Astrocyte-like Cells: Potential for Restorative Treatment in Parkinson’s Disease. J. Mol. Neurosci. 2009, 39, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Filous, A.R.; Miller, J.H.; Coulson-Thomas, Y.M.; Horn, K.P.; Alilain, W.J.; Silver, J. Immature Astrocytes Promote CNS Axonal Regeneration When Combined with Chondroitinase ABC. Dev. Neurobiol. 2010, 70, 826–841. [Google Scholar] [CrossRef] [PubMed]
- Gallizioli, M.; Miró-Mur, F.; Otxoa-de-Amezaga, A.; Cugota, R.; Salas-Perdomo, A.; Justicia, C.; Brait, V.H.; Ruiz-Jaén, F.; Arbaizar-Rovirosa, M.; Pedragosa, J.; et al. Dendritic Cells and Microglia Have Non-Redundant Functions in the Inflamed Brain with Protective Effects of Type 1 CDCs. Cell Rep. 2020, 33, 108291. [Google Scholar] [CrossRef]
- Qarin, S.; Howlett, S.K.; Jones, J.L.; Barker, R.A. The Immunogenicity of Midbrain Dopaminergic Neurons and the Implications for Neural Grafting Trials in Parkinson’s Disease. Neuronal Signal. 2021, 5, NS20200083. [Google Scholar] [CrossRef]
- Brundin, P.; Barker, R.A.; Parmar, M. Neural Grafting in Parkinson’s Disease Problems and Possibilities. Prog. Brain Res. 2010, 184, 265–294. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of Host-Derived α Synuclein to Grafted Dopaminergic Neurons in Rat. Neurobiol. Dis. 2011, 43, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-J.; Oh, S.-M.; Kwon, O.-C.; Wulansari, N.; Lee, H.-S.; Chang, M.-Y.; Lee, E.; Sun, W.; Lee, S.-E.; Chang, S.; et al. Cografting Astrocytes Improves Cell Therapeutic Outcomes in a Parkinson’s Disease Model. J. Clin. Investig. 2017, 128, 463–482. [Google Scholar] [CrossRef]
- Wei, Z.-Y.D.; Shetty, A.K. Treating Parkinson’s Disease by Astrocyte Reprogramming: Progress and Challenges. Sci. Adv. 2021, 7, eabg3198. [Google Scholar] [CrossRef]
- Ghasemi-Kasman, M.; Hajikaram, M.; Baharvand, H.; Javan, M. MicroRNA-Mediated In Vitro and In Vivo Direct Conversion of Astrocytes to Neuroblasts. PLoS ONE 2015, 10, e0127878. [Google Scholar] [CrossRef]
- Steiner, J.A.; Angot, E.; Brundin, P. A Deadly Spread: Cellular Mechanisms of α-Synuclein Transfer. Cell Death Differ. 2011, 18, 1425–1433. [Google Scholar] [CrossRef]
- Herrington, T.M.; Cheng, J.J.; Eskandar, E.N. Mechanisms of Deep Brain Stimulation. J. Neurophysiol. 2016, 115, 19–38. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Sun, Y.; Dettmer, U. Astrocytes in Parkinson’s Disease: From Role to Possible Intervention. Cells 2023, 12, 2336. https://doi.org/10.3390/cells12192336
Wang T, Sun Y, Dettmer U. Astrocytes in Parkinson’s Disease: From Role to Possible Intervention. Cells. 2023; 12(19):2336. https://doi.org/10.3390/cells12192336
Chicago/Turabian StyleWang, Tianyou, Yingqi Sun, and Ulf Dettmer. 2023. "Astrocytes in Parkinson’s Disease: From Role to Possible Intervention" Cells 12, no. 19: 2336. https://doi.org/10.3390/cells12192336