
Increasing Ciliary ARL13B Expression Drives Active and Inhibitor-Resistant Smoothened and GLI into Glioma Primary Cilia

 , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot
2.3. Bulk RNA Sequencing
2.4. Immunocytochemistry
2.5. Data Analysis
3. Results
3.1. ARL13B+ Cilia Are Present in GBM Biopsies and Recently Derived Patient Cell Lines
3.2. Increasing ARL13B Expression Correlates with an Increase in SMO and GLI2 and More Aggressive Glioma
3.3. Increasing a GFP-Tagged, Functional ARL13B in Glioma Cilia Elongates Them and Promotes SMO Accumulation
3.4. Increasing ARL13B Expression Promotes Abnormal Ciliary Tip Accumulation of Activated SMO
3.5. ARL13B-Induced Accumulation of Ciliary SMO and GLI2 Is Resistant to Potent SMO Inhibitors
3.6. ARL13B:GFP-Induced Changes in Cilia Are Not Associated with Canonical Downstream SHH Pathway Activation
3.7. Glioma Cilia Overexpressing WT, but Not T35N ARL13B, Display Reduced Ciliary INPP5e
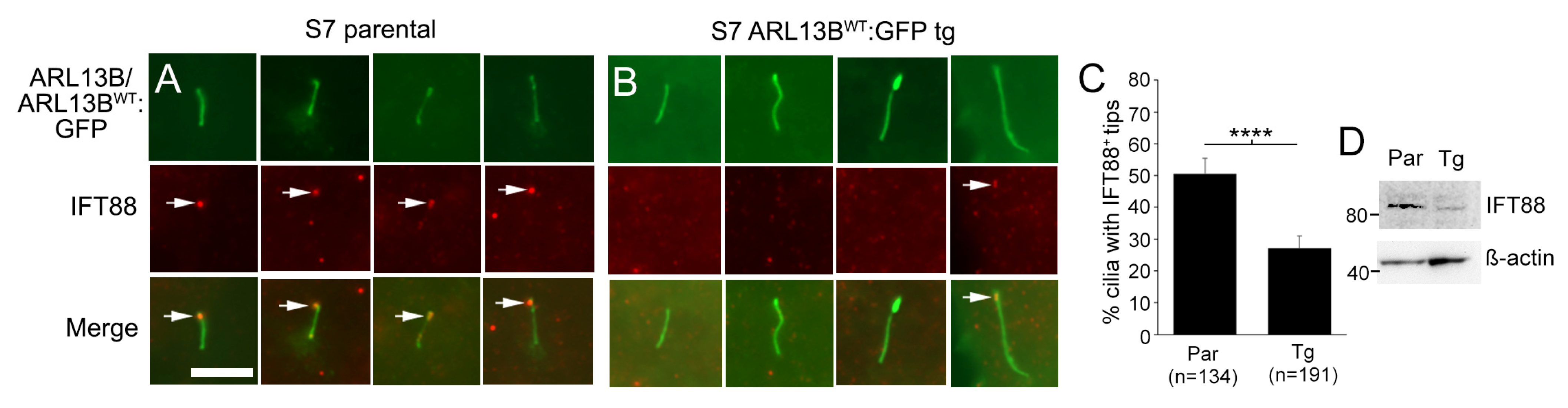
3.8. Glioma Cilia Overexpressing ARL13BWT:GFP Display Reduced IFT88
4. Discussion
4.1. ARL13B-Driven Increases in Ciliary SMO: Failed Retrieval of Activated SMO or a Resistance Mechanism in Glioma?
4.2. Are Glioma Cilia Non-Responsive or Responding Differently to SHH/SMO?
4.3. What Does High Expression of ARL13B, IFT88, and SMO in TCGA Signify?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Osborn, A.G.; Louis, D.N.; Poussaint, T.Y.; Linscott, L.L.; Salzman, K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. AJNR Am. J. Neuroradiol. 2022, 43, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Jooma, R.; Waqas, M.; Khan, I. Diffuse low-grade glioma—Changing concepts in diagnosis and management: A review. Asian J. Neurosurg. 2019, 14, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Aiman, W.; Gasalberti, D.P.; Rayi, A. Low Grade Gliomas. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Stupp, R.; Mason, W.P.; van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Alvarez-Satta, M.; Matheu, A. Primary cilium and glioblastoma. Ther. Adv. Med. Oncol. 2018, 10, 1758835918801169. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Han, Y.G.; Alvarez-Buylla, A. Role of primary cilia in brain development and cancer. Curr. Opin. Neurobiol. 2010, 20, 58–67. [Google Scholar] [CrossRef]
- Liu, H.; Kiseleva, A.A.; Golemis, E.A. Ciliary signalling in cancer. Nat. Rev. Cancer 2018, 18, 511–524. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol. 2007, 170, 347–355. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gruber Filbin, M.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef]
- Xu, Q.; Yuan, X.; Liu, G.; Black, K.L.; Yu, J.S. Hedgehog signaling regulates brain tumor-initiating cell proliferation and portends shorter survival for patients with PTEN-coexpressing glioblastomas. Stem Cells 2008, 26, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Goranci-Buzhala, G.; Mariappan, A.; Ricci-Vitiani, L.; Josipovic, N.; Pacioni, S.; Gottardo, M.; Ptok, J.; Schaal, H.; Callaini, G.; Rajalingam, K.; et al. Cilium induction triggers differentiation of glioma stem cells. Cell Rep. 2021, 36, 109656. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Ugartemendia, G.; Futch, H.; Griffith, B.; Breunig, J.J.; De Leon, G.; Mitchell, D.A.; Semple-Rowland, S.; et al. Disruption of KIF3A in patient-derived glioblastoma cells: Effects on ciliogenesis, hedgehog sensitivity, and tumorigenesis. Oncotarget 2016, 7, 7029–7043. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Dutra-Clarke, M.; Breunig, J.J.; Sarkisian, M.R. Glioma cell proliferation is enhanced in the presence of tumor-derived cilia vesicles. Cilia 2018, 7, 6. [Google Scholar] [CrossRef]
- Lee, D.; Gimple, R.C.; Wu, X.; Prager, B.C.; Qiu, Z.; Wu, Q.; Daggubati, V.; Mariappan, A.; Gopalakrishnan, J.; Sarkisian, M.R.; et al. Superenhancer activation of KLHDC8A drives glioma ciliation and hedgehog signaling. J. Clin. Investig. 2023, 133, e163592. [Google Scholar] [CrossRef]
- Sarkisian, M.R.; Siebzehnrubl, D.; Hoang-Minh, L.; Deleyrolle, L.; Silver, D.J.; Siebzehnrubl, F.A.; Guadiana, S.M.; Srivinasan, G.; Semple-Rowland, S.; Harrison, J.K.; et al. Detection of primary cilia in human glioblastoma. J. Neurooncol. 2014, 117, 15–24. [Google Scholar] [CrossRef]
- Wei, L.; Ma, W.; Cai, H.; Peng, S.P.; Tian, H.B.; Wang, J.F.; Gao, L.; He, J.P. Inhibition of Ciliogenesis Enhances the Cellular Sensitivity to Temozolomide and Ionizing Radiation in Human Glioblastoma Cells. Biomed. Environ. Sci. 2022, 35, 419–436. [Google Scholar] [CrossRef]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Hoang-Minh, L.B.; Tian, J.; Cheng, A.; Basrai, R.; Kalaria, N.; Lebowitz, J.J.; Khoshbouei, H.; Deleyrolle, L.P.; Sarkisian, M.R. HDAC6 Signaling at Primary Cilia Promotes Proliferation and Restricts Differentiation of Glioma Cells. Cancers 2021, 13, 1644. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, X.; Wang, T.; Xu, L.; Zhai, Z.; Wu, H.; Deng, L.; Lu, Q.; Chen, Z.; Yang, X.; et al. ARL13B promotes angiogenesis and glioma growth by activating VEGFA-VEGFR2 signaling. Neuro Oncol. 2023, 25, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Savchuk, S.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 2021, 144, 1230–1246. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef]
- Larkins, C.E.; Aviles, G.D.; East, M.P.; Kahn, R.A.; Caspary, T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol. Biol. Cell 2011, 22, 4694–4703. [Google Scholar] [CrossRef]
- Mariani, L.E.; Bijlsma, M.F.; Ivanova, A.I.; Suciu, S.K.; Kahn, R.A.; Caspary, T. Arl13b regulates Shh signaling from both inside and outside the cilium. Mol. Biol. Cell 2016, 27, 3780–3790. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.E14. [Google Scholar] [CrossRef]
- Kinnebrew, M.; Iverson, E.J.; Patel, B.B.; Pusapati, G.V.; Kong, J.H.; Johnson, K.A.; Luchetti, G.; Eckert, K.M.; McDonald, J.G.; Covey, D.F.; et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. eLife 2019, 8, e50051. [Google Scholar] [CrossRef]
- Raleigh, D.R.; Sever, N.; Choksi, P.K.; Sigg, M.A.; Hines, K.M.; Thompson, B.M.; Elnatan, D.; Jaishankar, P.; Bisignano, P.; Garcia-Gonzalo, F.R.; et al. Cilia-Associated Oxysterols Activate Smoothened. Mol. Cell 2018, 72, 316–327.E5. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Ocbina, P.J.; Anderson, K.V. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 2009, 94, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.; Reiter, J.F. A central region of Gli2 regulates its localization to the primary cilium and transcriptional activity. J. Cell Sci. 2014, 127, 1500–1510. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Lu, H.; Toh, M.T.; Narasimhan, V.; Thamilselvam, S.K.; Choksi, S.P.; Roy, S. A function for the Joubert syndrome protein Arl13b in ciliary membrane extension and ciliary length regulation. Dev. Biol. 2015, 397, 225–236. [Google Scholar] [CrossRef]
- Gigante, E.D.; Taylor, M.R.; Ivanova, A.A.; Kahn, R.A.; Caspary, T. ARL13B regulates Sonic hedgehog signaling from outside primary cilia. eLife 2020, 9, e50434. [Google Scholar] [CrossRef]
- Gursel, D.B.; Connell-Albert, Y.S.; Tuskan, R.G.; Anastassiadis, T.; Walrath, J.C.; Hawes, J.J.; Amlin-Van Schaick, J.C.; Reilly, K.M. Control of proliferation in astrocytoma cells by the receptor tyrosine kinase/PI3K/AKT signaling axis and the use of PI-103 and TCN as potential anti-astrocytoma therapies. Neuro Oncol. 2011, 13, 610–621. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef]
- Hothi, P.; Martins, T.J.; Chen, L.; Deleyrolle, L.; Yoon, J.G.; Reynolds, B.; Foltz, G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 2012, 3, 1124–1136. [Google Scholar] [CrossRef]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Moser, J.J.; Fritzler, M.J.; Rattner, J.B. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 2009, 9, 448. [Google Scholar] [CrossRef]
- Zalenski, A.A.; Majumder, S.; De, K.; Venere, M. An interphase pool of KIF11 localizes at the basal bodies of primary cilia and a reduction in KIF11 expression alters cilia dynamics. Sci. Rep. 2020, 10, 13946. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Caspary, T.; Seyfried, N.T.; Duong, D.M.; West, A.B.; Liu, Z.; Kahn, R.A. Biochemical characterization of purified mammalian ARL13B protein indicates that it is an atypical GTPase and ARL3 guanine nucleotide exchange factor (GEF). J. Biol. Chem. 2017, 292, 11091–11108. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Walsh, C.T.; McMahon, A.P. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc. Natl. Acad. Sci. USA 2009, 106, 2623–2628. [Google Scholar] [CrossRef]
- Desai, P.B.; Stuck, M.W.; Lv, B.; Pazour, G.J. Ubiquitin links smoothened to intraflagellar transport to regulate Hedgehog signaling. J. Cell Biol. 2020, 219, e201912104. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.O.; Campbell, V.T.; Harari, J.A.; Tibbitts, T.T.; Proctor, J.L.; Whitebread, N.; Conley, J.M.; White, K.F.; Kutok, J.L.; Read, M.A.; et al. Impact of the Smoothened inhibitor, IPI-926, on smoothened ciliary localization and Hedgehog pathway activity. PLoS ONE 2014, 9, e90534. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.; Ena, S.; Van Sande, J.; de Kerchove d’Exaerde, A.; Schurmans, S.; Schiffmann, S.N. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev. Cell 2015, 34, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Phua, S.C.; Roberson, E.C.; Garcia, G., 3rd; Abedin, M.; Schurmans, S.; Inoue, T.; Reiter, J.F. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev. Cell 2015, 34, 400–409. [Google Scholar] [CrossRef]
- Qiu, H.; Fujisawa, S.; Nozaki, S.; Katoh, Y.; Nakayama, K. Interaction of INPP5E with ARL13B is essential for its ciliary membrane retention but dispensable for its ciliary entry. Biol. Open 2021, 10, bio057653. [Google Scholar] [CrossRef]
- Humbert, M.C.; Weihbrecht, K.; Searby, C.C.; Li, Y.; Pope, R.M.; Sheffield, V.C.; Seo, S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 19691–19696. [Google Scholar] [CrossRef]
- Dyson, J.M.; Conduit, S.E.; Feeney, S.J.; Hakim, S.; DiTommaso, T.; Fulcher, A.J.; Sriratana, A.; Ramm, G.; Horan, K.A.; Gurung, R.; et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J. Cell Biol. 2017, 216, 247–263. [Google Scholar] [CrossRef]
- Schembs, L.; Willems, A.; Hasenpusch-Theil, K.; Cooper, J.D.; Whiting, K.; Burr, K.; Bostrand, S.M.K.; Selvaraj, B.T.; Chandran, S.; Theil, T. The ciliary gene INPP5E confers dorsal telencephalic identity to human cortical organoids by negatively regulating Sonic hedgehog signaling. Cell Rep. 2022, 39, 110811. [Google Scholar] [CrossRef]
- Munoz-Estrada, J.; Ferland, R.J. Ahi1 promotes Arl13b ciliary recruitment, regulates Arl13b stability and is required for normal cell migration. J. Cell Sci. 2019, 132, jcs230680. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Taulman, P.D.; Haycraft, C.J.; Balkovetz, D.F.; Yoder, B.K. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol. Biol. Cell 2001, 12, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Nager, A.R.; Goldstein, J.S.; Herranz-Perez, V.; Portran, D.; Ye, F.; Garcia-Verdugo, J.M.; Nachury, M.V. An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell 2017, 168, 252–263.E14. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Nager, A.R.; Nachury, M.V. BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 2018, 217, 1847–1868. [Google Scholar] [CrossRef]
- Shao, J.; Xu, L.; Chen, L.; Lu, Q.; Xie, X.; Shi, W.; Xiong, H.; Shi, C.; Huang, X.; Mei, J.; et al. Arl13b promotes gastric tumorigenesis by regulating Smo trafficking and activation of the hedgehog signaling pathway. Cancer Res. 2017, 77, 4000–4013. [Google Scholar] [CrossRef]
- Milenkovic, L.; Scott, M.P.; Rohatgi, R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 2009, 187, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.K.; Fei, D.L.; Schilling, N.S.; Ahmed, Y.F.; Hwa, J.; Robbins, D.J. G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature 2008, 456, 967–970. [Google Scholar] [CrossRef]
- Riobo, N.A.; Saucy, B.; Dilizio, C.; Manning, D.R. Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. USA 2006, 103, 12607–12612. [Google Scholar] [CrossRef]
- Shen, F.; Cheng, L.; Douglas, A.E.; Riobo, N.A.; Manning, D.R. Smoothened is a fully competent activator of the heterotrimeric G protein G(i). Mol. Pharmacol. 2013, 83, 691–697. [Google Scholar] [CrossRef]
- Findakly, S.; Choudhury, A.; Daggubati, V.; Pekmezci, M.; Lang, U.E.; Raleigh, D.R. Meningioma cells express primary cilia but do not transduce ciliary Hedgehog signals. Acta Neuropathol. Commun. 2020, 8, 114. [Google Scholar] [CrossRef]
- Spann, A.L.; Yuan, K.; Goliwas, K.F.; Steg, A.D.; Kaushik, D.D.; Kwon, Y.J.; Frost, A.R. The presence of primary cilia in cancer cells does not predict responsiveness to modulation of smoothened activity. Int. J. Oncol. 2015, 47, 269–279. [Google Scholar] [CrossRef]
- Jenks, A.D.; Vyse, S.; Wong, J.P.; Kostaras, E.; Keller, D.; Burgoyne, T.; Shoemark, A.; Tsalikis, A.; de la Roche, M.; Michaelis, M.; et al. Primary cilia mediate diverse kinase inhibitor resistance mechanisms in cancer. Cell Rep. 2018, 23, 3042–3055. [Google Scholar] [CrossRef] [PubMed]
- Delaval, B.; Bright, A.; Lawson, N.D.; Doxsey, S. The cilia protein IFT88 is required for spindle orientation in mitosis. Nat. Cell Biol. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Tian, J.; Ulm, B.S.; Mallinger, J.C.; Khoshbouei, H.; Deleyrolle, L.P.; Sarkisian, M.R. Tumor Treating Fields Suppression of Ciliogenesis Enhances Temozolomide Toxicity. Front. Oncol. 2022, 12, 837589. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host Species | Dilution | Manufacturer/Catalogue # |
|---|---|---|---|
| ADP ribosylation factor 13B (ARL13B) | Rabbit | 1:3000 | Proteintech; Cat #17711-1-AP |
| ADP ribosylation factor 13B (ARL13B) | Mouse | 1:3000 | Abcam; Cat #AB136648 |
| Acetylated alpha Tubulin (aaTub) | Mouse | 1:3000 | Sigma; Cat #T6793 |
| Beta-actin | Mouse | 1:10,000 | Sigma; Cat #A5316 |
| Flag | Mouse | 1:5000 | Sigma; Cat #F-9291 |
| Gamma-tubulin (gTub) | Mouse | 1:3000 | Sigma; Cat #T6557 |
| Green fluorescent protein (GFP) | Chicken | 1:5000 | Abcam; Cat #13970 |
| GLI2 | Goat | 1:500 | R&D Systems; Cat #AF3635 |
| Inositol polyphosphate 5-phosphatase (INPP5E) | Rabbit | 1:1000 | Proteintech; Cat #17797-1-AP |
| Intraflagellar transport 88 (IFT88) | Rabbit | 1:1000 | Proteintech; Cat #13967-1-AP |
| Pericentriolar material 1 (PCM1) | Rabbit | 1:3000 | Bethyl; Cat #A301150A |
| Smoothened (SMO) | Rabbit | 1:1000 | Abcam; Cat #AB38686 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, P.; Tian, J.; Mallinger, J.C.; Ling, D.; Deleyrolle, L.P.; McIntyre, J.C.; Caspary, T.; Breunig, J.J.; Sarkisian, M.R. Increasing Ciliary ARL13B Expression Drives Active and Inhibitor-Resistant Smoothened and GLI into Glioma Primary Cilia. Cells 2023, 12, 2354. https://doi.org/10.3390/cells12192354
Shi P, Tian J, Mallinger JC, Ling D, Deleyrolle LP, McIntyre JC, Caspary T, Breunig JJ, Sarkisian MR. Increasing Ciliary ARL13B Expression Drives Active and Inhibitor-Resistant Smoothened and GLI into Glioma Primary Cilia. Cells. 2023; 12(19):2354. https://doi.org/10.3390/cells12192354
Chicago/Turabian StyleShi, Ping, Jia Tian, Julianne C. Mallinger, Dahao Ling, Loic P. Deleyrolle, Jeremy C. McIntyre, Tamara Caspary, Joshua J. Breunig, and Matthew R. Sarkisian. 2023. "Increasing Ciliary ARL13B Expression Drives Active and Inhibitor-Resistant Smoothened and GLI into Glioma Primary Cilia" Cells 12, no. 19: 2354. https://doi.org/10.3390/cells12192354