


MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction

 , ,
, ,  ,
,  ,
,  , ,
, ,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
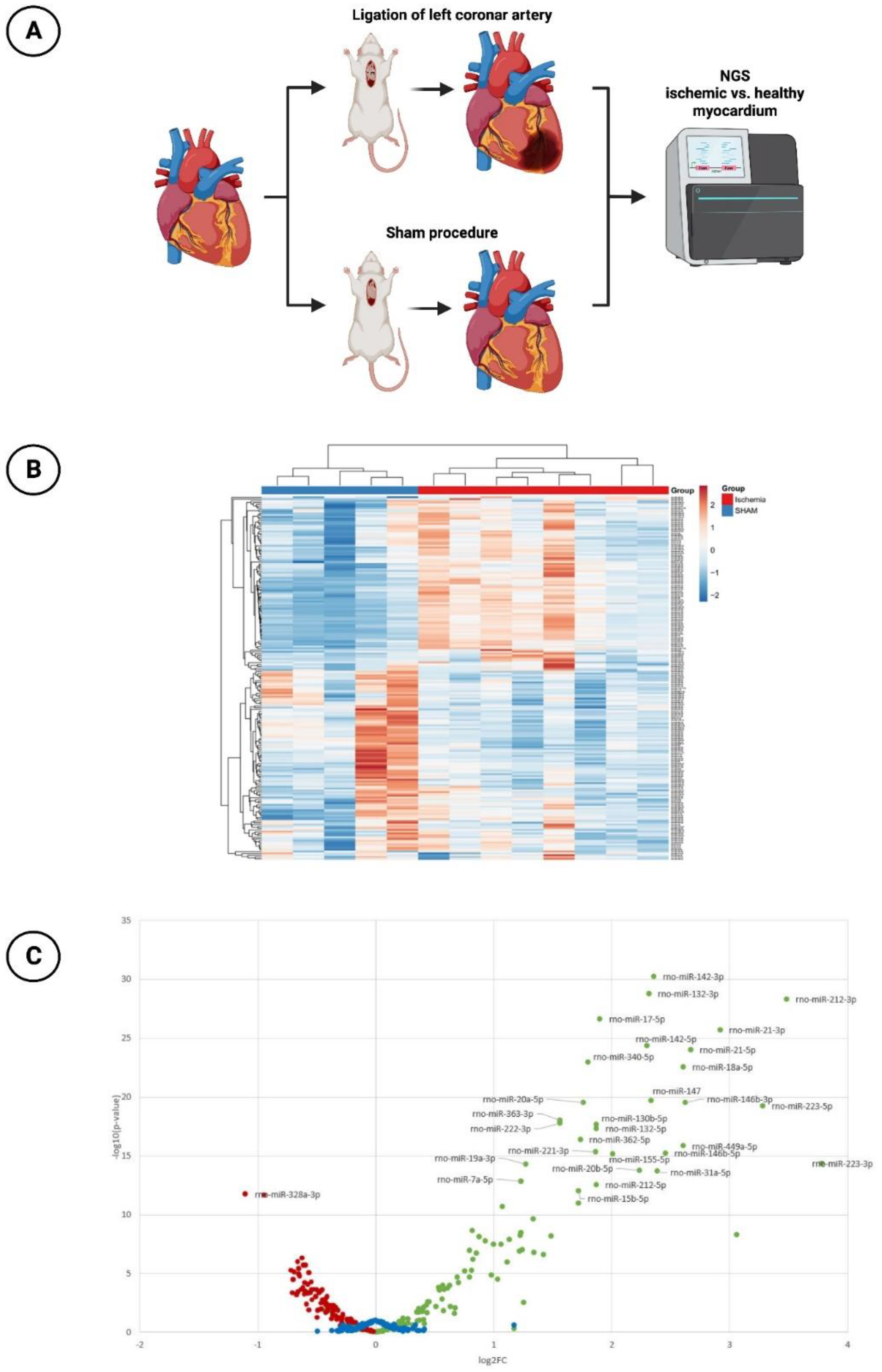
2.1. Experimental Rat Model
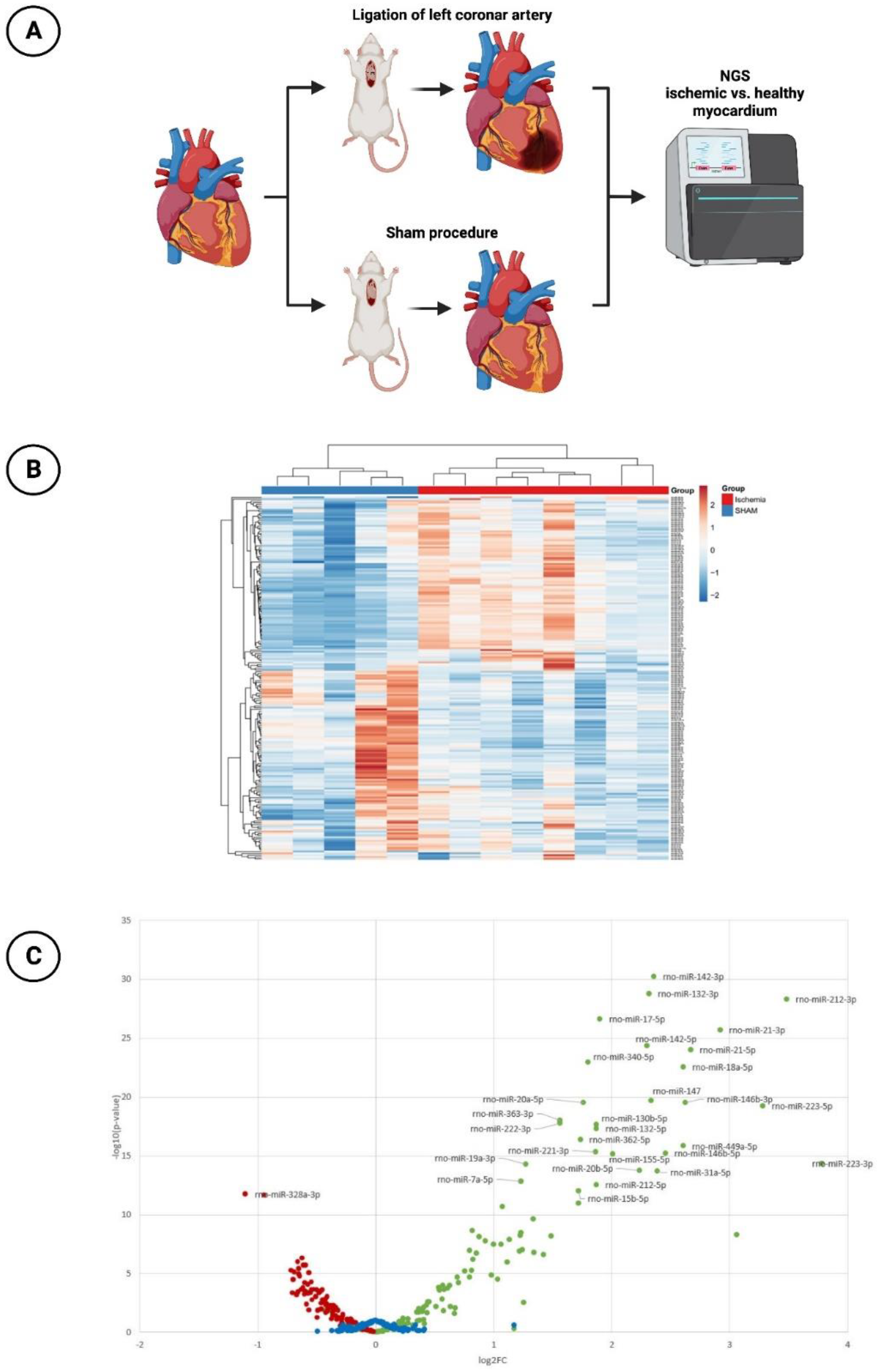
2.1.1. Induction of Acute Myocardial Infarction
2.1.2. MiR Expression with Next-Generation Sequencing (NGS)
Total RNA Extraction from Tissue
Small RNA Sequencing
Small RNA Sequencing Data Analysis
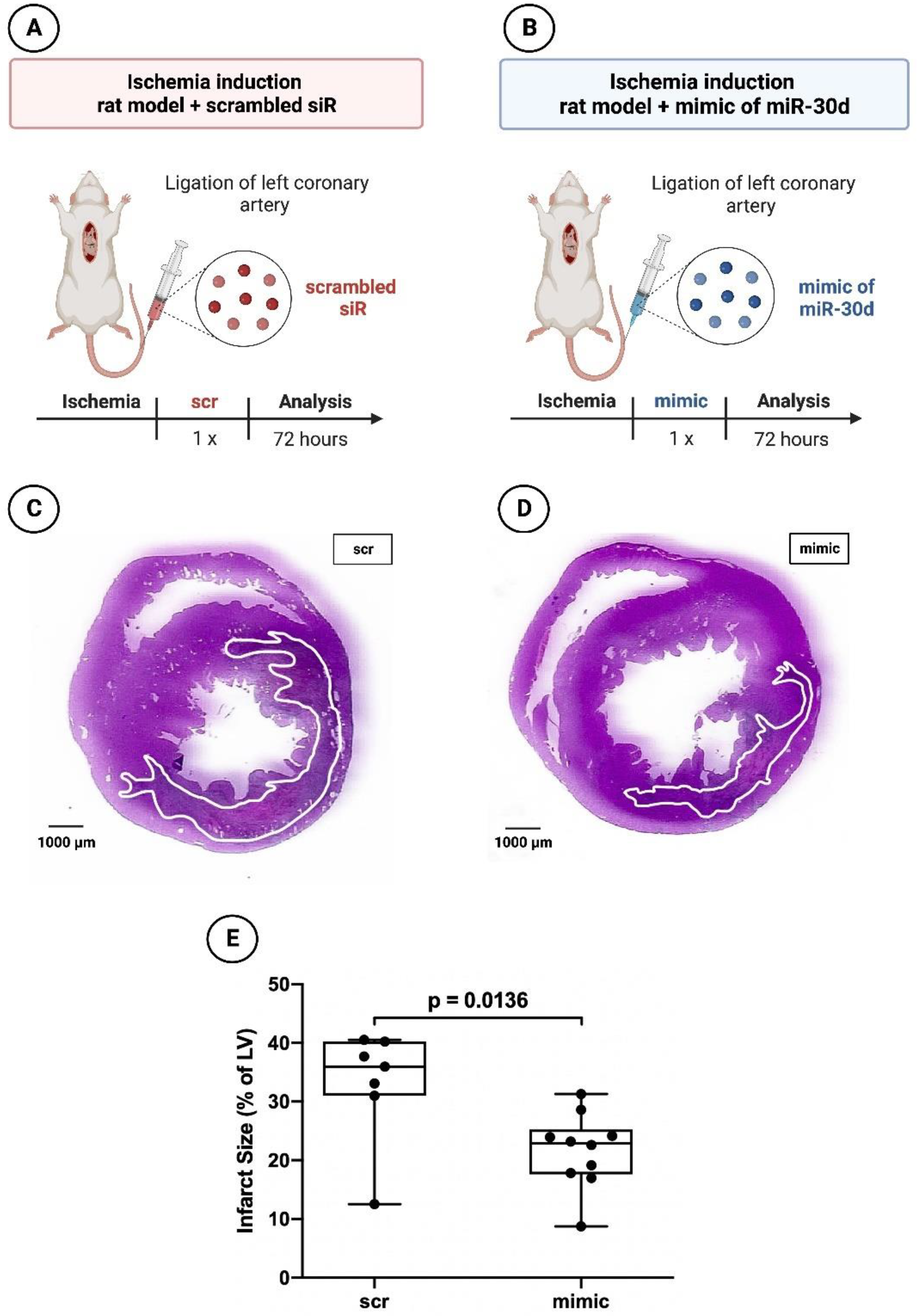
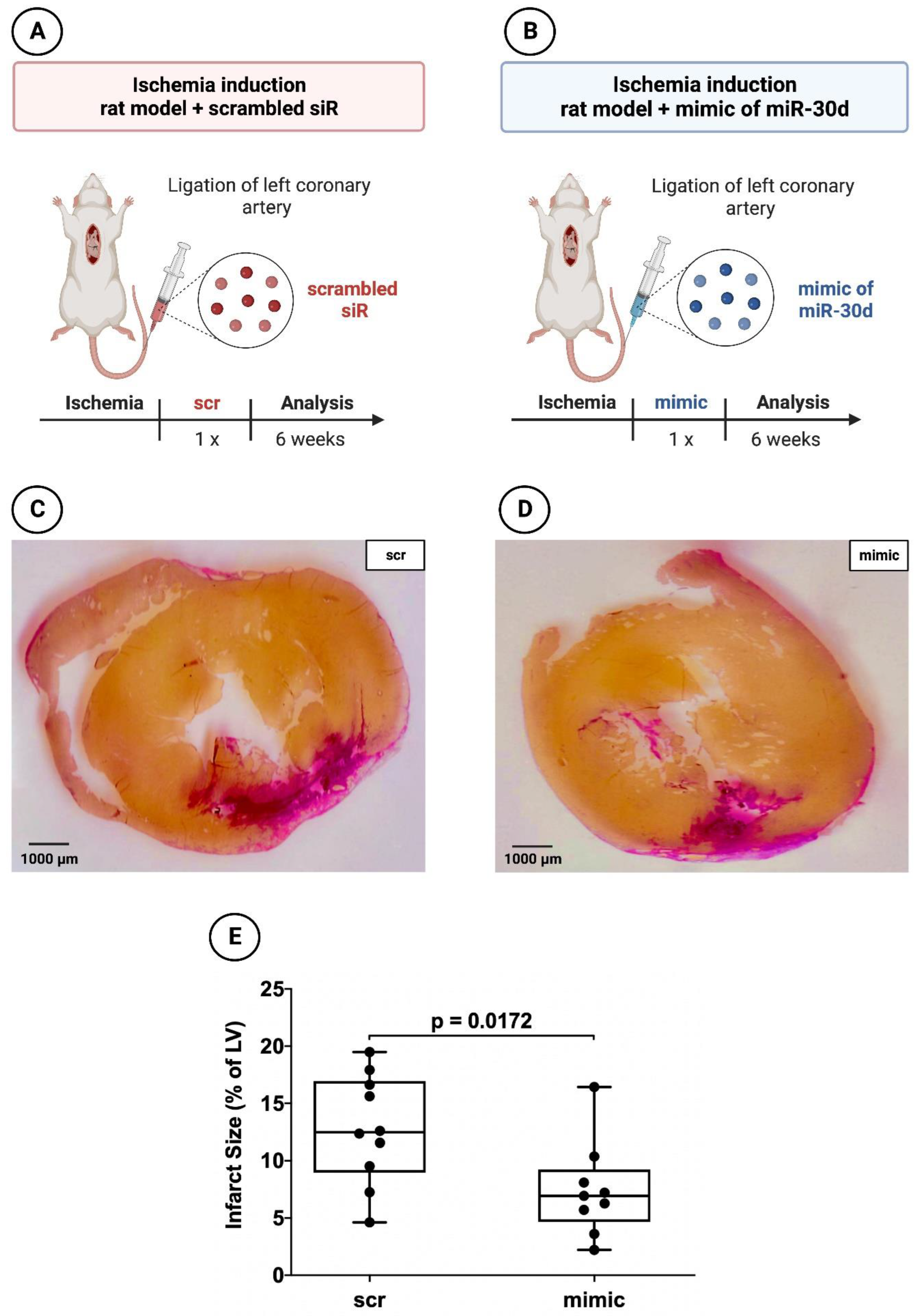
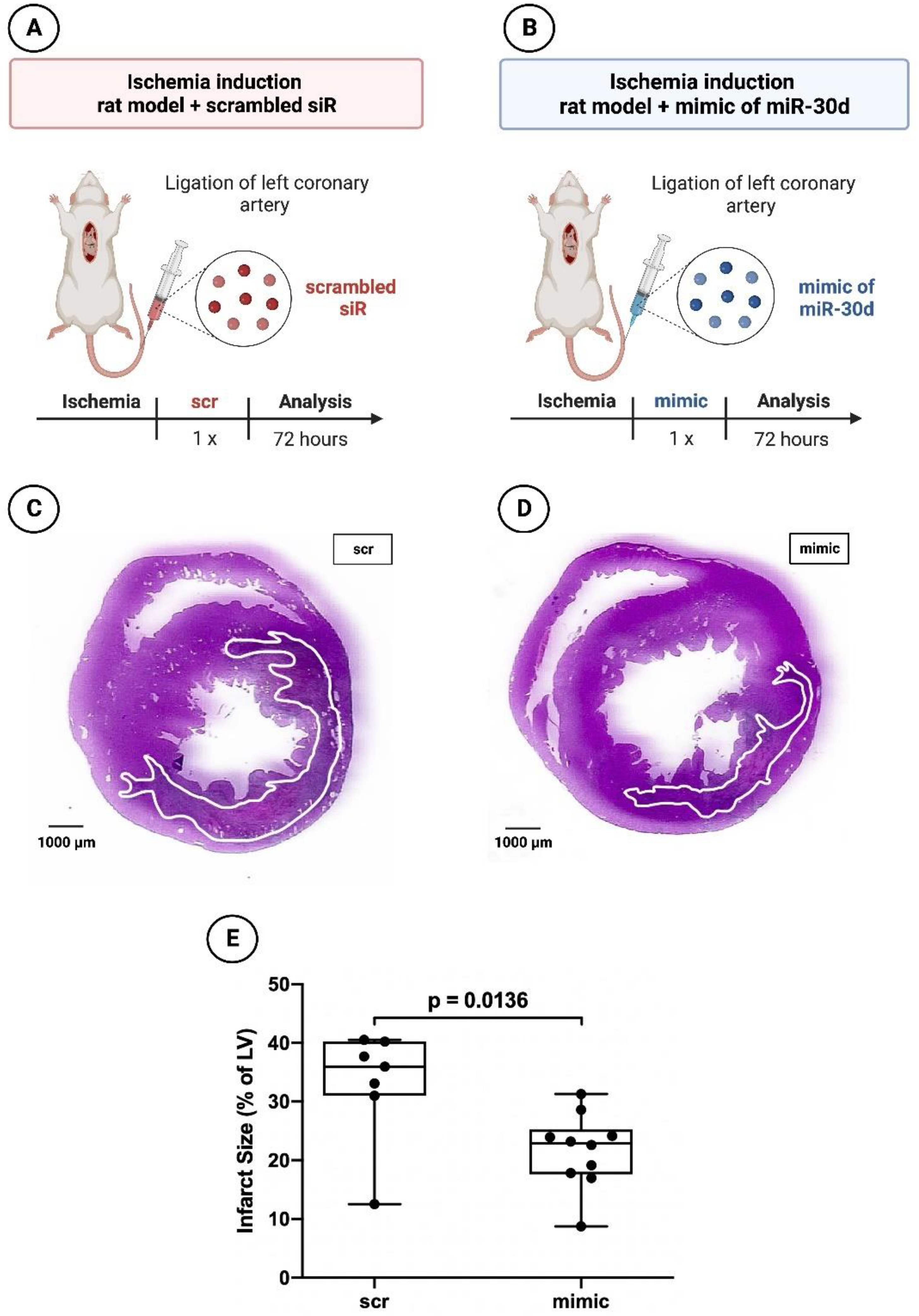
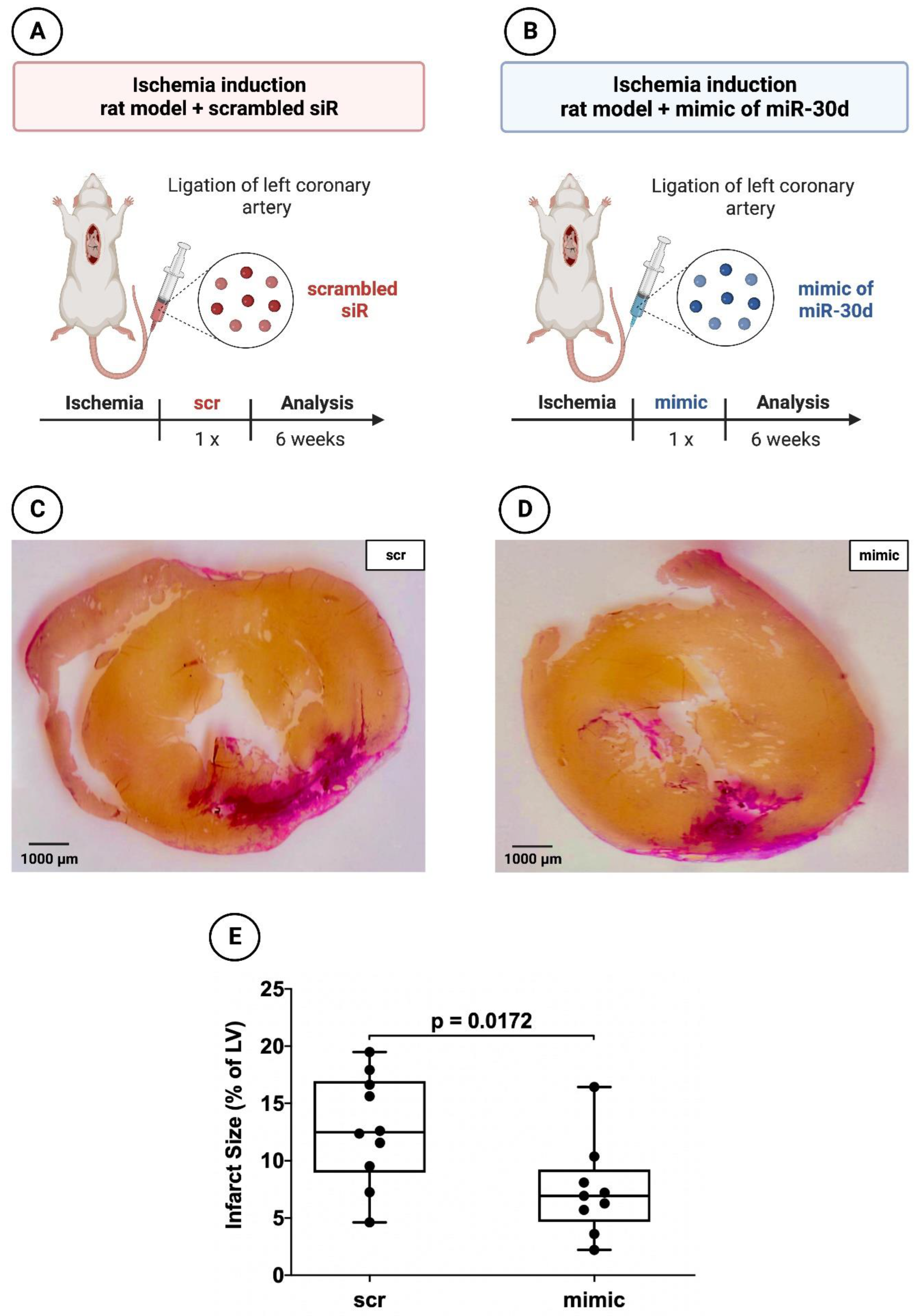
2.1.3. Histology and Planimetry
2.2. Human Umbilical Cord Endothelial Cells (HUVECs)
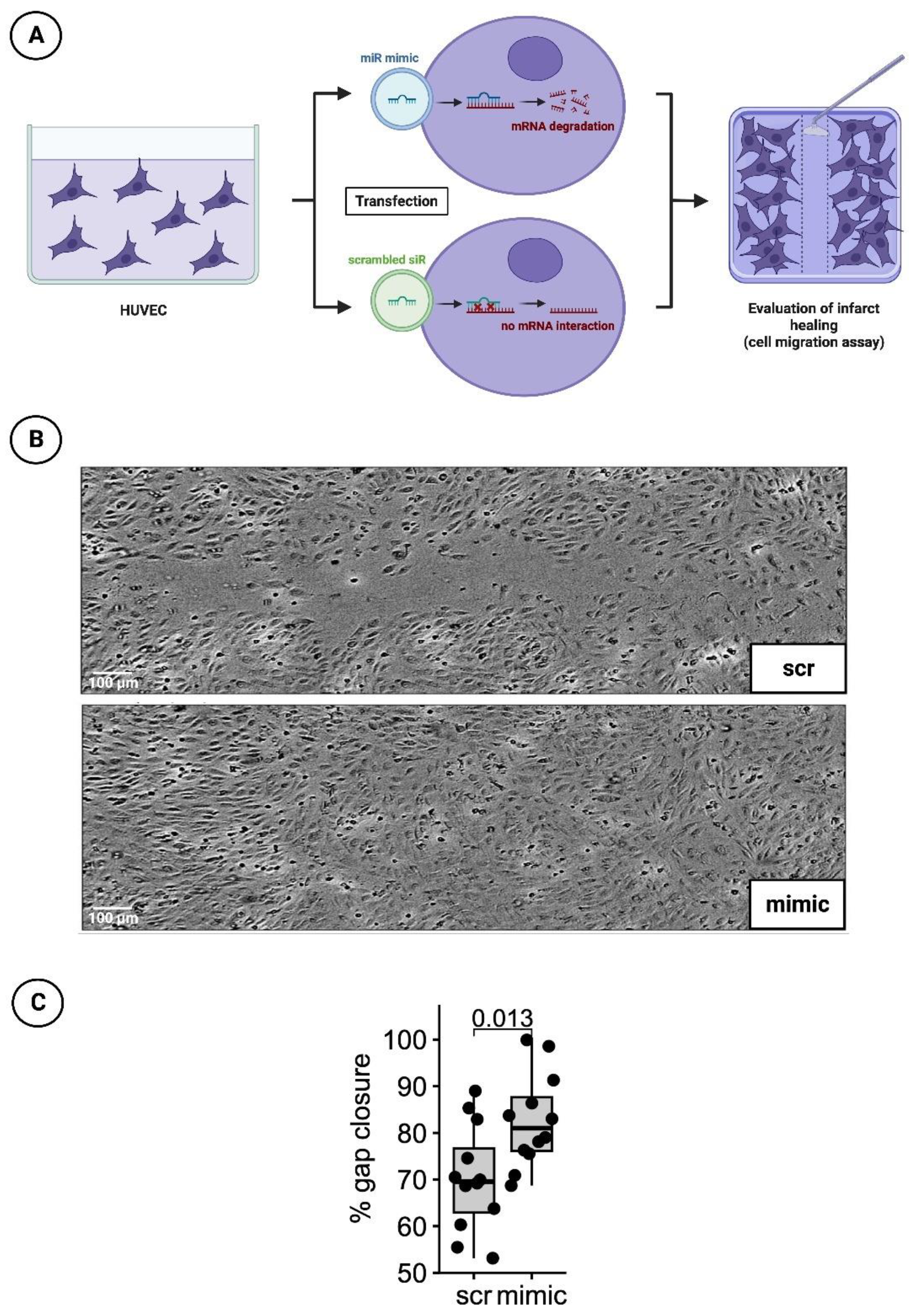
2.2.1. Cultivation and Transfection with Nanoparticles of the miR Mimic or Scrambled siR
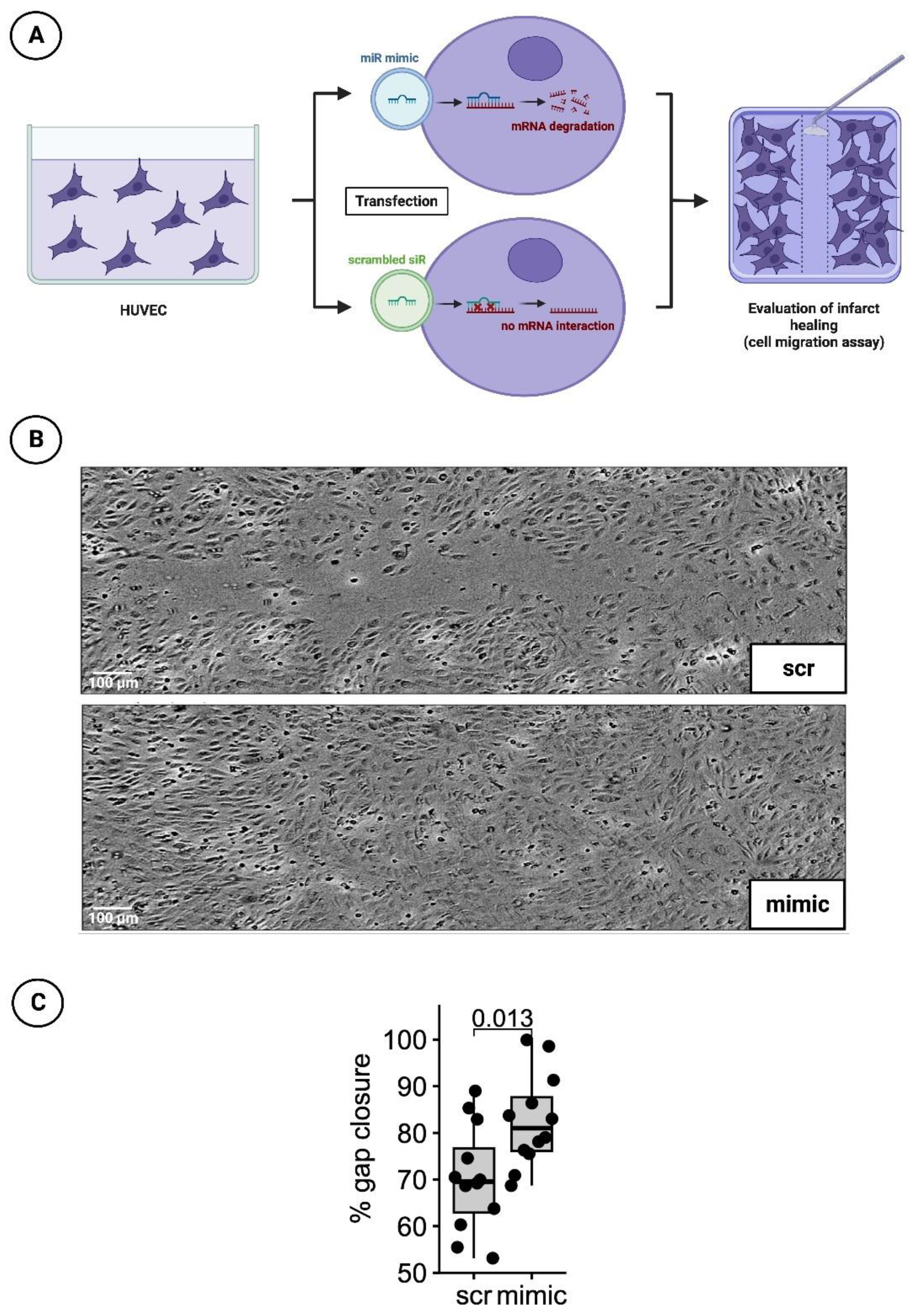
2.2.2. Detection of Infarct Healing via Cell Migration Scratch Assay
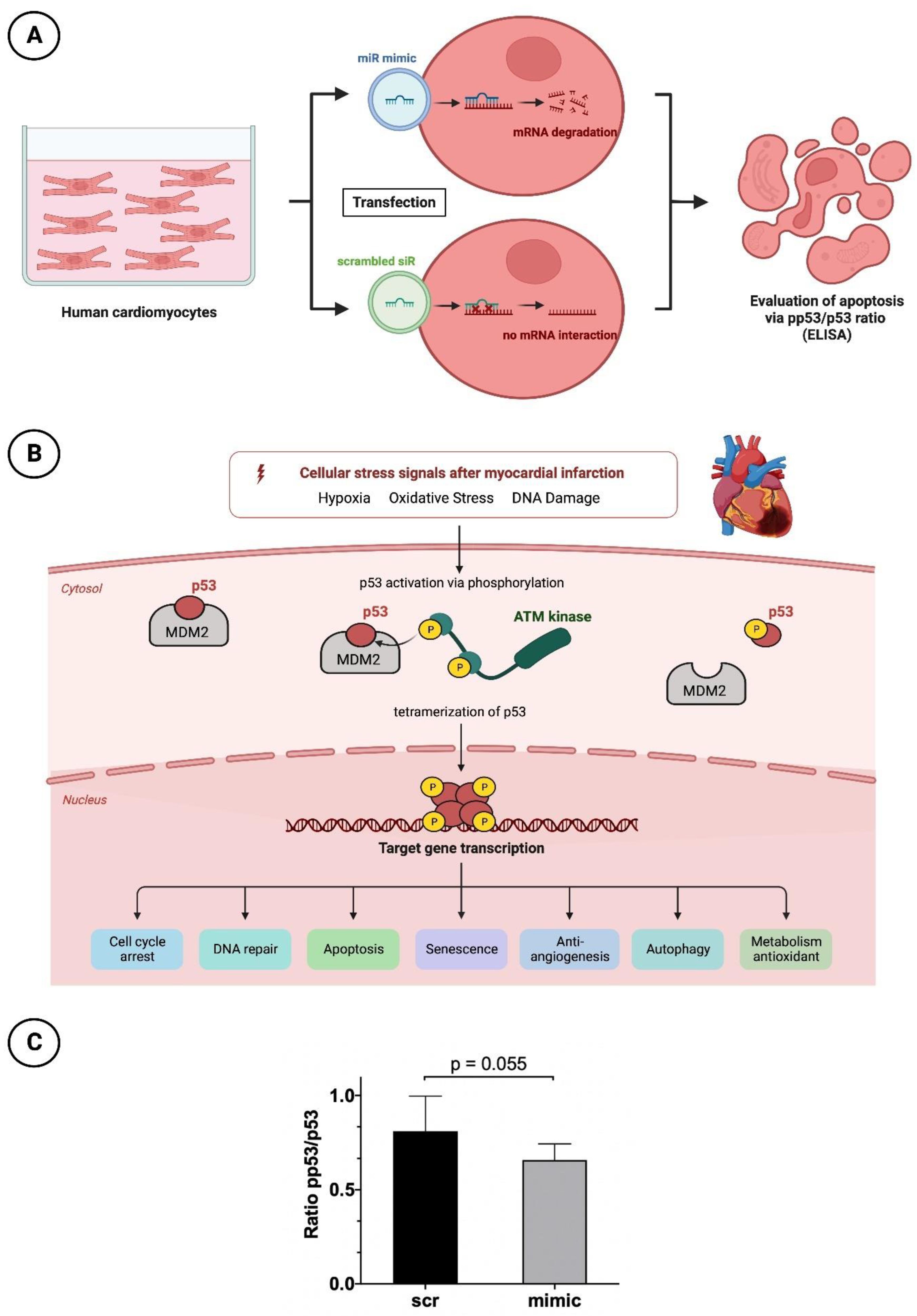
2.3. Human Cardiomyocytes (HCMs)
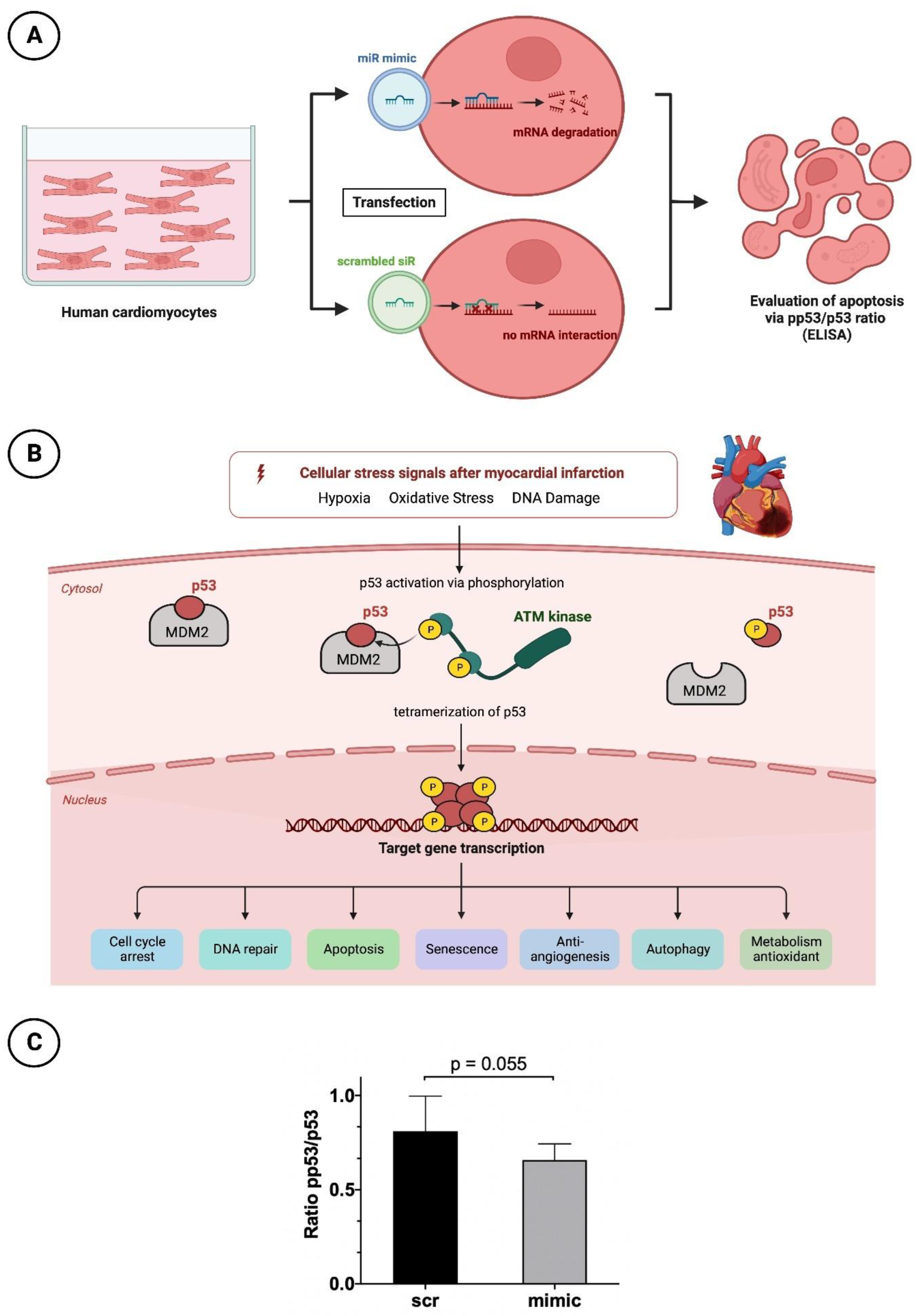
2.3.1. Cultivation and Transfection with Nanoparticles of the miR Mimic or Scrambled siR
2.3.2. Apoptosis Testing by p53 and pp53 ELISA
2.4. Statistical Analysis
3. Results
3.1. miR-30d-5p with Significant Downregulation in the Animal Model of Ischemic CMP
3.2. Significant Reduction in Infarct areal Size in Animals Treated with the Mimic of miR-30d-5p after 72 h and after 6 Weeks
3.3. Significantly Faster Gap Closure in Cell Migration Scratch Assay of HUVECs Transfected with the Mimic of miR-30d-5p
3.4. Reduced Apoptotic Rate in HCMs Transfected with the Mimic of miR-30d-5p
4. Discussion
4.1. Anti-Remodeling Therapy—A Therapeutic Option to Reduce the Extent of Ischemia
4.2. What Do miRs Offer Regarding other Therapeutic Options in the Course of MI?
4.3. MiR-30d-5p and its Role in Ischemic CMP
4.4. Resistance to Ischemia, Anti-Apoptosis and Proliferation—Mechanisms of Action through Therapeutic Increase in miR-30d-5p
5. Limitation
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhandari, B.; Quintanilla Rodriguez, B.S.; Masood, W. Ischemic Cardiomyopathy. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Nowbar, A.N.; Gitto, M.; Howard, J.P.; Francis, D.P.; Al-Lamee, R. Mortality From Ischemic Heart Disease. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005375. [Google Scholar] [CrossRef] [PubMed]
- Abreu, L.M. Time is Muscle. Arq. Bras. Cardiol. 2019, 112, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Brinks, J.; Fowler, A.; Franklin, B.A.; Dulai, J. Lifestyle Modification in Secondary Prevention: Beyond Pharmacotherapy. Am. J. Lifestyle Med. 2016, 11, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Yarnell, J.W.; Sweetnam, P.M.; Rumley, A.; Lowe, G.D. Lifestyle and hemostatic risk factors for ischemic heart disease: The Caerphilly Study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Çitaku, H.; Miftari, R.; Stubljar, D.; Krasniqi, X. Size of Acute Myocardial Infarction Correlates with Earlier Time of Initiation of Reperfusion Therapy with Cardiac Perfusion Scintigraphy: A National Single-Center Study. Med. Sci. Monit. Basic Res. 2021, 27, e933214-1–e933214-6. [Google Scholar] [CrossRef]
- Wagdy, H.M.; Christian, T.F. Determinants of infarct size in acute myocardial infarction in patients treated with reperfusion therapy. Curr. Opin. Cardiol. 1996, 11, 369–377. [Google Scholar] [CrossRef]
- Simonis, G.; Strasser, R.H.; Ebner, B. Reperfusion injury in acute myocardial infarction. Crit. Care 2012, 16 (Suppl. S2), A22. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef]
- Gosselin, H.; Qi, X.; Rouleau, J.L. Correlation between cardiac remodelling, function, and myocardial contractility in rat hearts 5 weeks after myocardial infarction. Can. J. Physiol. Pharmacol. 1998, 76, 53–62. [Google Scholar] [CrossRef]
- Kong, A.S.; Lai, K.S.; Lim, S.E.; Sivalingam, S.; Loh, J.Y.; Maran, S. miRNA in Ischemic Heart Disease and Its Potential as Biomarkers: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 9001. [Google Scholar] [CrossRef] [PubMed]
- Song, M.A.; Paradis, A.N.; Gay, M.S.; Shin, J.; Zhang, L. Differential expression of microRNAs in ischemic heart disease. Drug Discov. Today 2015, 20, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Caroli, A.; Cardillo, M.T.; Galea, R.; Biasucci, L.M. Potential therapeutic role of microRNAs in ischemic heart disease. J. Cardiol. 2013, 61, 315–320. [Google Scholar] [CrossRef]
- Colpaert RM, W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef]
- Peterlin, A.; Počivavšek, K.; Petrovič, D.; Peterlin, B. The Role of microRNAs in Heart Failure: A Systematic Review. Front. Cardiovasc. Med. 2020, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Dong, Y.H.; Du, W.; Shi, C.Y.; Wang, K.; Tariq, M.A.; Wang, J.X.; Li, P.F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef]
- Zhai, C.; Li, R.; Hou, K.; Chen, J.; Alzogool, M.; Hu, Y.; Zhang, J.; Zhang, Y.; Wang, L.; Zhang, R.; et al. Value of Blood-Based microRNAs in the Diagnosis of Acute Myocardial Infarction: A Systematic Review and Meta-Analysis. Front. Physiol. 2020, 11, 691. [Google Scholar] [CrossRef]
- Scărlătescu, A.I.; Micheu, M.M.; Popa-Fotea, N.M.; Dorobanțu, M. MicroRNAs in Acute ST Elevation Myocardial Infarction-A New Tool for Diagnosis and Prognosis: Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 4799. [Google Scholar] [CrossRef]
- Yang, X.; Du, X.; Ma, K.; Li, G.; Liu, Z.; Rong, W.; Miao, H.; Zhu, F.; Cui, Q.; Wu, S.; et al. Circulating miRNAs Related to Long-term Adverse Cardiovascular Events in STEMI Patients: A Nested Case-Control Study. Can. J. Cardiol. 2021, 37, 77–85. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. TIG 2022, 38, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohen, S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tang, X.; Liu, X.; Cui, X.; Lian, M.; Zhao, M.; Peng, H.; Han, X. Targeted miR-21 loaded liposomes for acute myocardial infarction. J. Mater. Chem. B 2020, 8, 10384–10391. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function After Myocardial Infarction. Circ. Res. 2017, 120, 1298–1304. [Google Scholar] [CrossRef]
- Wei, Y.; Peng, S.; Wu, M.; Sachidanandam, R.; Tu, Z.; Zhang, S.; Falce, C.; Sobie, E.A.; Lebeche, D.; Zhao, Y. Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell Res. 2014, 24, 278–292. [Google Scholar] [CrossRef]
- Fiedler, J.; Jazbutyte, V.; Kirchmaier, B.C.; Gupta, S.K.; Lorenzen, J.; Hartmann, D.; Galuppo, P.; Kneitz, S.; Pena, J.T.; Sohn-Lee, C.; et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 2011, 124, 720–730. [Google Scholar] [CrossRef]
- Foinquinos, A.; Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Gyöngyösi, M.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020, 11, 633. [Google Scholar] [CrossRef]
- Hinkel, R.; Batkai, S.; Bähr, A.; Bozoglu, T.; Straub, S.; Borchert, T.; Viereck, J.; Howe, A.; Hornaschewitz, N.; Oberberger, L.; et al. AntimiR-132 Attenuates Myocardial Hypertrophy in an Animal Model of Percutaneous Aortic Constriction. J. Am. Coll. Cardiol. 2021, 77, 2923–2935. [Google Scholar] [CrossRef]
- Gonçalves, I.F.; Acar, E.; Costantino, S.; Szabo, P.L.; Hamza, O.; Tretter, E.V.; Klein, K.U.; Trojanek, S.; Abraham, D.; Paneni, F.; et al. Epigenetic modulation of tenascin C in the heart: Implications on myocardial ischemia, hypertrophy and metabolism. J. Hypertens. 2019, 37, 1861–1870. [Google Scholar] [CrossRef]
- Pilz, P.M.; Hamza, O.; Gidlöf, O.; Gonçalves, I.F.; Tretter, E.V.; Trojanek, S.; Abraham, D.; Heber, S.; Haller, P.M.; Podesser, B.K.; et al. Remote ischemic perconditioning attenuates adverse cardiac remodeling and preserves left ventricular function in a rat model of reperfused myocardial infarction. Int. J. Cardiol. 2019, 285, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, H.; Kogel, A.; Kalwa, H.; Weirauch, U.; Aigner, A. Therapeutic miR-506-3p Replacement in Pancreatic Carcinoma Leads to Multiple Effects including Autophagy, Apoptosis, Senescence, and Mitochondrial Alterations In Vitro and In Vivo. Biomedicines 2022, 10, 1692. [Google Scholar] [CrossRef] [PubMed]
- Ewe, A.; Przybylski, S.; Burkhardt, J.; Janke, A.; Appelhans, D.; Aigner, A. A novel tyrosine-modified low molecular weight polyethylenimine (P10Y) for efficient siRNA delivery in vitro and in vivo. J. Control. Release Off. J. Control. Release Soc. 2016, 230, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Werth, S.; Urban-Klein, B.; Dai, L.; Höbel, S.; Grzelinski, M.; Bakowsky, U.; Czubayko, F.; Aigner, A. A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J. Control. Release Off. J. Control. Release Soc. 2006, 112, 257–270. [Google Scholar] [CrossRef]
- Zhupanyn, P.; Ewe, A.; Büch, T.; Malek, A.; Rademacher, P.; Müller, C.; Reinert, A.; Jaimes, Y.; Aigner, A. Extracellular vesicle (ECV)-modified polyethylenimine (PEI) complexes for enhanced siRNA delivery in vitro and in vivo. J. Control. Release Off. J. Control. Release Soc. 2020, 319, 63–76. [Google Scholar] [CrossRef]
- Lichtenauer, M.; Schreiber, C.; Jung, C.; Beer, L.; Mangold, A.; Gyöngyösi, M.; Podesser, B.K.; Ankersmit, H.J. Myocardial infarct size measurement using geometric angle calculation. Eur. J. Clin. Investig. 2014, 44, 160–167. [Google Scholar] [CrossRef]
- Lichtenauer, M.; Mildner, M.; Werba, G.; Beer, L.; Hoetzenecker, K.; Baumgartner, A.; Hasun, M.; Nickl, S.; Mitterbauer, A.; Zimmermann, M.; et al. Anti-thymocyte globulin induces neoangiogenesis and preserves cardiac function after experimental myocardial infarction. PLoS ONE 2012, 7, e52101. [Google Scholar] [CrossRef]
- Wernly, B.; Paar, V.; Aigner, A.; Pilz, P.M.; Podesser, B.K.; Förster, M.; Jung, C.; Hofbauer, J.P.; Tockner, B.; Wimmer, M.; et al. Anti-CD3 Antibody Treatment Reduces Scar Formation in a Rat Model of Myocardial Infarction. Cells 2020, 9, 295. [Google Scholar] [CrossRef]
- Bostjancic, E.; Zidar, N.; Glavac, D. MicroRNA microarray expression profiling in human myocardial infarction. Dis. Markers 2009, 27, 255–268. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 769. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, X.R.; Han, X. MicroRNA-146b induces the PI3K/Akt/NF-κB signaling pathway to reduce vascular inflammation and apoptosis in myocardial infarction by targeting PTEN. Exp. Ther. Med. 2019, 17, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, J.; Guo, M.; Hao, M. MiR-223-3p affects myocardial inflammation and apoptosis following myocardial infarction via targeting FBXW7. J. Thorac. Dis. 2022, 14, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, M.; Roubina, E. Drugs for left ventricular remodeling in heart failure. Am. J. Cardiol. 2005, 96, 10L–18L. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef]
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. BioMed Res. Int. 2017, 2017, 1278436. [Google Scholar] [CrossRef] [PubMed]
- Topkara, V.K.; Mann, D.L. Clinical applications of miRNAs in cardiac remodeling and heart failure. Pers. Med. 2010, 7, 531–548. [Google Scholar] [CrossRef]
- Maries, L.; Marian, C.; Sosdean, R.; Goanta, F.; Sirbu, I.O.; Anghel, A. MicroRNAs-The Heart of Post-Myocardial Infarction Remodeling. Diagnostics 2021, 11, 1675. [Google Scholar] [CrossRef]
- Boon, R.A.; Dimmeler, S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, H. Myocardial Infarction: The Protective Role of MiRNAs in Myocardium Pathology. Front. Cardiovasc. Med. 2021, 8, 631817. [Google Scholar] [CrossRef]
- Xiao, J.; Gao, R.; Bei, Y.; Zhou, Q.; Zhou, Y.; Zhang, H.; Jin, M.; Wei, S.; Wang, K.; Xu, X.; et al. Circulating miR-30d Predicts Survival in Patients with Acute Heart Failure. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 865–874. [Google Scholar] [CrossRef]
- Melman, Y.F.; Shah, R.; Danielson, K.; Xiao, J.; Simonson, B.; Barth, A.; Chakir, K.; Lewis, G.D.; Lavender, Z.; Truong, Q.A.; et al. Circulating MicroRNA-30d Is Associated with Response to Cardiac Resynchronization Therapy in Heart Failure and Regulates Cardiomyocyte Apoptosis: A Translational Pilot Study. Circulation 2015, 131, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Shi, P.; Han, X.; Chen, T.; Tang, H.; Wang, J. Diagnostic value of miR-30d-5p and miR-125b-5p in acute myocardial infarction. Mol. Med. Rep. 2016, 14, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, T.; Mickus, R.; Cereskevicius, D.; Macas, A. Value of Serum miR-23a, miR-30d, and miR-146a Biomarkers in ST-Elevation Myocardial Infarction. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, Z.; Zhu, X.; Xu, W.; Yuan, W.; Yang, T.; Jin, B.; Yan, Y.; Chen, R.; Wang, S.; et al. Targeting miR-30d reverses pathological cardiac hypertrophy. EBioMedicine 2022, 81, 104108. [Google Scholar] [CrossRef]
- Jiang, M.; Wang, H.; Jin, M.; Yang, X.; Ji, H.; Jiang, Y.; Zhang, H.; Wu, F.; Wu, G.; Lai, X.; et al. Exosomes from MiR-30d-5p-ADSCs Reverse Acute Ischemic Stroke-Induced, Autophagy-Mediated Brain Injury by Promoting M2 Microglial/Macrophage Polarization. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 864–878. [Google Scholar] [CrossRef]
- Zhao, F.; Qu, Y.; Zhu, J.; Zhang, L.; Huang, L.; Liu, H.; Li, S.; Mu, D. miR-30d-5p Plays an Important Role in Autophagy and Apoptosis in Developing Rat Brains After Hypoxic-Ischemic Injury. J. Neuropathol. Exp. Neurol. 2017, 76, 709–719. [Google Scholar] [CrossRef]
- Tang, S.; Wang, Y.; Ma, T.; Lu, S.; Huang, K.; Li, Q.; Wu, M.; Yang, H.; Zhong, J. MiR-30d inhibits cardiomyocytes autophagy promoting ferroptosis after myocardial infarction. Panminerva Med. 2020. advance online publication. [Google Scholar] [CrossRef]
- Kim, J.O.; Park, J.H.; Kim, T.; Hong, S.E.; Lee, J.Y.; Nho, K.J.; Cho, C.; Kim, Y.S.; Kang, W.S.; Ahn, Y.; et al. A novel system-level approach using RNA-sequencing data identifies miR-30-5p and miR-142a-5p as key regulators of apoptosis in myocardial infarction. Sci. Rep. 2018, 8, 14638. [Google Scholar] [CrossRef]
- Yu, H.; Lin, X.; Wang, F.; Zhang, B.; Wang, W.; Shi, H.; Zou, B.; Zhao, J. Proliferation inhibition and the underlying molecular mechanisms of microRNA-30d in renal carcinoma cells. Oncol. Lett. 2014, 7, 799–804. [Google Scholar] [CrossRef]
- Xu, X.; Zong, K.; Wang, X.; Dou, D.; Lv, P.; Zhang, Z.; Li, H. miR-30d suppresses proliferation and invasiveness of pancreatic cancer by targeting the SOX4/PI3K-AKT axis and predicts poor outcome. Cell Death Dis. 2021, 12, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, J.; Zhao, J.; Bai, J. Mir-30d suppresses cell proliferation of colon cancer cells by inhibiting cell autophagy and promoting cell apoptosis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317703984. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | log2(FC) | p-Value |
|---|---|---|
| rno-miRNA-223-3p | 3.78 | 4.64 × 10−15 |
| rno-miRNA-212-3p | 3.48 | 4.49 × 10−29 |
| rno-miRNA-223-5p | 3.28 | 5.71 × 10−20 |
| rno-miRNA-3473 | 3.06 | 5.27 × 10−09 |
| rno-miRNA-21-3p | 2.92 | 1.96 × 10−26 |
| rno-miRNA-21-5p | 2.67 | 9.28 × 10−25 |
| rno-miRNA-146b-3p | 2.62 | 2.99 × 10−20 |
| rno-miRNA-449a-5p | 2.61 | 1.47 × 10−16 |
| rno-miRNA-18a-5p | 2.61 | 2.71 × 10−23 |
| rno-miRNA-146b-5p | 2.46 | 5.72 × 10−16 |
| rno-miRNA-31a-5p | 2.39 | 2.07 × 10−14 |
| rno-miRNA-142-3p | 2.36 | 6.02 × 10−31 |
| rno-miRNA-147 | 2.34 | 2.09 × 10−20 |
| rno-miRNA-132-3p | 2.32 | 1.69 × 10−29 |
| rno-miRNA-142-5p | 2.30 | 4.14 × 10−25 |
| rno-miRNA-20b-5p | 2.24 | 1.82 × 10−14 |
| rno-miRNA-155-5p | 2.01 | 7.11 × 10−16 |
| rno-miRNA-17-5p | 1.90 | 2.47 × 10−27 |
| rno-miRNA-212-5p | 1.87 | 2.75 × 10−13 |
| rno-miRNA-221-3p | 1.86 | 4.37 × 10−16 |
| miRNA | log2(FC) | p-Value |
|---|---|---|
| rno-miRNA-328a-3p | −1.10 | 1.79 × 10−12 |
| rno-miR-125a-5p | −0.94 | 2.04 × 10−12 |
| rno-miR-874-3p | −0.70 | 4.85 × 10−04 |
| rno-miRNA-133-5p | −0.69 | 3.46 × 10−05 |
| rno-miRNA-100-5p | −0.68 | 2.92 × 10−08 |
| rno-miR-325-5p | −0.67 | 6.82 × 10−04 |
| rno-miRNA-351-5p | −0.66 | 1.14 × 10−06 |
| rno-miRNA-30d-5p | −0.65 | 4.27 × 10−06 |
| rno-miRNA-128-3p | −0.65 | 1.29 × 10−05 |
| rno-miRNA-1-3p | −0.64 | 1.72 × 10−05 |
| rno-miR-150-3p | −0.63 | 2.49 × 10−04 |
| rno-miRNA-26a-5p | −0.62 | 5.16 × 10−07 |
| rno- miR- 30e- 5p | −0.62 | 1.64 × 10−04 |
| rno-miR-125b-5p | −0.61 | 2.00 × 10−06 |
| rno-miRNA-22-3p | −0.60 | 3.12 × 10−04 |
| rno-miRNA-181d-5p | −0.59 | 2.24 × 10−06 |
| rno-miR-208b-3p | −0.59 | 4.62 × 10−03 |
| rno-miR-22-5p | −0.58 | 5.24 × 10−03 |
| rno-miR-145-3p | −0.56 | 9.70 × 10−06 |
| rno-miRNA-664a-3p | −0.55 | 4.76 × 10−04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boxhammer, E.; Paar, V.; Wernly, B.; Kiss, A.; Mirna, M.; Aigner, A.; Acar, E.; Watzinger, S.; Podesser, B.K.; Zauner, R.; et al. MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction. Cells 2023, 12, 2369. https://doi.org/10.3390/cells12192369
Boxhammer E, Paar V, Wernly B, Kiss A, Mirna M, Aigner A, Acar E, Watzinger S, Podesser BK, Zauner R, et al. MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction. Cells. 2023; 12(19):2369. https://doi.org/10.3390/cells12192369
Chicago/Turabian StyleBoxhammer, Elke, Vera Paar, Bernhard Wernly, Attila Kiss, Moritz Mirna, Achim Aigner, Eylem Acar, Simon Watzinger, Bruno K. Podesser, Roland Zauner, and et al. 2023. "MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction" Cells 12, no. 19: 2369. https://doi.org/10.3390/cells12192369
APA StyleBoxhammer, E., Paar, V., Wernly, B., Kiss, A., Mirna, M., Aigner, A., Acar, E., Watzinger, S., Podesser, B. K., Zauner, R., Wally, V., Ablinger, M., Hackl, M., Hoppe, U. C., & Lichtenauer, M. (2023). MicroRNA-30d-5p—A Potential New Therapeutic Target for Prevention of Ischemic Cardiomyopathy after Myocardial Infarction. Cells, 12(19), 2369. https://doi.org/10.3390/cells12192369