
PAK1 and Therapy Resistance in Melanoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Therapy of Metastatic Melanoma: An Unmet Need Remains despite the Recent Progress
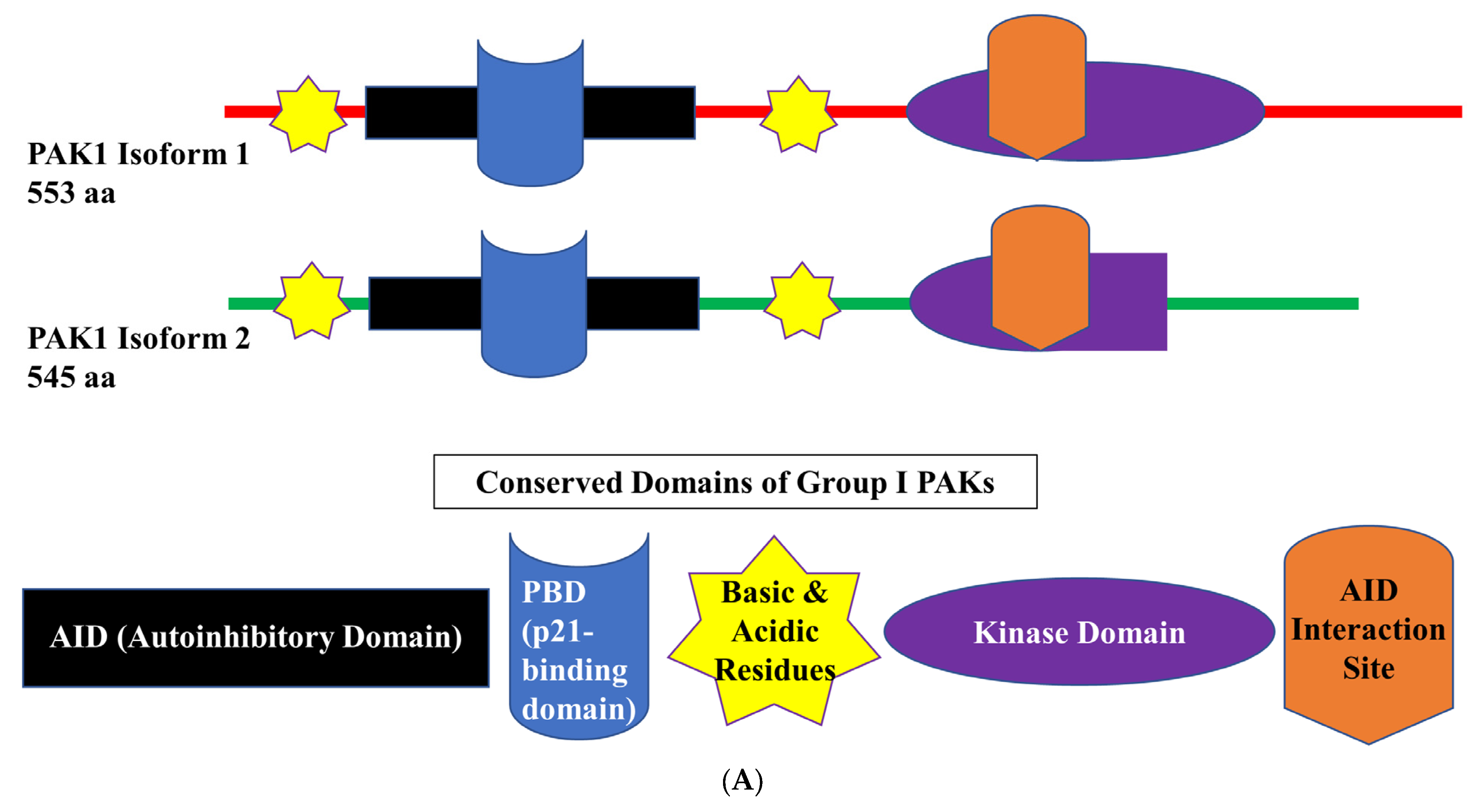
2. PAK1 Structure and Expression in Melanoma

3. PAK1 and Genotoxic Therapy in Melanoma
4. PAK1 and Targeted Therapies in Melanoma
5. PAK1 and Immunotherapy of Melanoma
6. Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Daniels, A.B. Continued Poor Survival in Metastatic Uveal Melanoma: Implications for Molecular Prognostication, Surveillance Imaging, Adjuvant Therapy, and Clinical Trials. JAMA Ophthalmol. 2018, 136, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.; Bellet, J.S.; Rubin, A.I.; Lipner, S.R. Adult and Pediatric Nail Unit Melanoma: Epidemiology, Diagnosis, and Treatment. Cells 2023, 12, 964. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Einhorn, L.H.; Meyers, M.L.; Saxman, S.; Destro, A.N.; Panageas, K.S.; Begg, C.B.; Agarwala, S.S.; Schuchter, L.M.; Ernstoff, M.S.; et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J. Clin. Oncol. 1999, 17, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.F.; Aamdal, S.; Grob, J.J.; Hauschild, A.; Mohr, P.; Bonerandi, J.J.; Weichenthal, M.; Neuber, K.; Bieber, T.; Gilde, K.; et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: A phase III study. J. Clin. Oncol. 2004, 22, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.K.; Kirkwood, J.M. Medical management of melanoma. Surg. Clin. N. Am. 2003, 83, 283–322. [Google Scholar] [CrossRef]
- Bajetta, E.; Del Vecchio, M.; Bernard-Marty, C.; Vitali, M.; Buzzoni, R.; Rixe, O.; Nova, P.; Aglione, S.; Taillibert, S.; Khayat, D. Metastatic melanoma: Chemotherapy. Semin. Oncol. 2002, 29, 427–445. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef]
- Caksa, S.; Baqai, U.; Aplin, A.E. The future of targeted kinase inhibitors in melanoma. Pharmacol. Ther. 2022, 239, 108200. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.F.; Choi, J.; Sosman, J. New Approaches to Targeted Therapy in Melanoma. Cancers 2023, 15, 3224. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.; Karapetyan, L.; Kirkwood, J.M. Immunotherapy in Melanoma: Recent Advances and Future Directions. Cancers 2023, 15, 1106. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet. Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Piulats, J.M.; Espinosa, E.; de la Cruz Merino, L.; Varela, M.; Alonso Carrion, L.; Martin-Algarra, S.; Lopez Castro, R.; Curiel, T.; Rodriguez-Abreu, D.; Redrado, M.; et al. Nivolumab Plus Ipilimumab for Treatment-Naive Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J. Clin. Oncol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results From a Single-Arm Phase II Study. J. Clin. Oncol. 2021, 39, 599–607. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Grebenova, D.; Holoubek, A.; Roselova, P.; Obr, A.; Brodska, B.; Kuzelova, K. PAK1, PAK1Delta15, and PAK2: Similarities, differences and mutual interactions. Sci. Rep. 2019, 9, 17171. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Si, W.; Liu, X.; He, L.; Ren, J.; Yang, Z.; Yang, J.; Li, W.; Liu, S.; Pei, F.; et al. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer 2017, 16, 175. [Google Scholar] [CrossRef]
- Chu, Y.; Elrod, N.; Wang, C.; Li, L.; Chen, T.; Routh, A.; Xia, Z.; Li, W.; Wagner, E.J.; Ji, P. Nudt21 regulates the alternative polyadenylation of Pak1 and is predictive in the prognosis of glioblastoma patients. Oncogene 2019, 38, 4154–4168. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Kandel, E.S. miRNAs: Little known mediators of oncogenesis. Semin. Cancer Biol. 2008, 18, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Strange, K.; Denton, J.; Nehrke, K. Ste20-type kinases: Evolutionarily conserved regulators of ion transport and cell volume. Physiology 2006, 21, 61–68. [Google Scholar] [CrossRef]
- Kichina, J.V.; Goc, A.; Al-Husein, B.; Somanath, P.R.; Kandel, E.S. PAK1 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 703–725. [Google Scholar] [CrossRef]
- Lei, M.; Lu, W.; Meng, W.; Parrini, M.C.; Eck, M.J.; Mayer, B.J.; Harrison, S.C. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 2000, 102, 387–397. [Google Scholar] [CrossRef]
- Strochlic, T.I.; Viaud, J.; Rennefahrt, U.E.; Anastassiadis, T.; Peterson, J.R. Phosphoinositides are essential coactivators for p21-activated kinase 1. Mol. Cell 2010, 40, 493–500. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.G.; Morris, S.; Dong, H.J.; Chernoff, J.; Bokoch, G.M. Regulation of human leukocyte p21-activated kinases through G protein—coupled receptors. Science 1995, 269, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Pirruccello, M.; Sondermann, H.; Pelton, J.G.; Pellicena, P.; Hoelz, A.; Chernoff, J.; Wemmer, D.E.; Kuriyan, J. A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J. Mol. Biol. 2006, 361, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, J.W.; Wang, Z.X. Structural insights into the autoactivation mechanism of p21-activated protein kinase. Structure 2011, 19, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.; Owen, D.; Chalk, P.A.; Lowe, P.N. Delineation of the Cdc42/Rac-binding domain of p21-activated kinase. Biochemistry 1998, 37, 7885–7891. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Reilly, A.M.; Daniels, R.H.; King, C.C.; Olivera, A.; Spiegel, S.; Knaus, U.G. A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J. Biol. Chem. 1998, 273, 8137–8144. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, biochemistry, and biology of PAK kinases. Gene 2017, 605, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Rane, C.K.; Minden, A. P21 activated kinase signaling in cancer. Semin. Cancer Biol. 2019, 54, 40–49. [Google Scholar] [CrossRef]
- Yao, D.; Li, C.; Rajoka, M.S.R.; He, Z.; Huang, J.; Wang, J.; Zhang, J. P21-Activated Kinase 1: Emerging biological functions and potential therapeutic targets in Cancer. Theranostics 2020, 10, 9741–9766. [Google Scholar] [CrossRef]
- Somanath, P.R.; Chernoff, J.; Cummings, B.S.; Prasad, S.M.; Homan, H.D. Targeting P21-Activated Kinase-1 for Metastatic Prostate Cancer. Cancers 2023, 15, 2236. [Google Scholar] [CrossRef]
- Babagana, M.; Johnson, S.; Slabodkin, H.; Bshara, W.; Morrison, C.; Kandel, E.S. P21-activated kinase 1 regulates resistance to BRAF inhibition in human cancer cells. Mol. Carcinog. 2017, 56, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.; Zuidervaart, W.; van Nieuwpoort, F.; Packer, L.; Jager, M.; Gruis, N.; Hayward, N. Increased p21-activated kinase-1 expression is associated with invasive potential in uveal melanoma. Melanoma Res. 2006, 16, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.C.; Jubb, A.M.; Jakubiak, D.; Zhou, W.; Rudolph, J.; Haverty, P.M.; Kowanetz, M.; Yan, Y.; Tremayne, J.; Lisle, R.; et al. P21-activated kinase 1 (PAK1) as a therapeutic target in BRAF wild-type melanoma. J. Natl. Cancer Inst. 2013, 105, 606–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Banik, I.; Shain, A.H.; Yeh, I.; Bastian, B.C. Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes. Genome Med. 2022, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Wilmott, J.S.; Johansson, P.A.; Nones, K.; Addala, V.; Mukhopadhyay, P.; Broit, N.; Amato, C.M.; Van Gulick, R.; Kazakoff, S.H.; et al. Whole-genome sequencing of acral melanoma reveals genomic complexity and diversity. Nat. Commun. 2020, 11, 5259. [Google Scholar] [CrossRef] [PubMed]
- Gumaste, P.V.; Fleming, N.H.; Silva, I.; Shapiro, R.L.; Berman, R.S.; Zhong, J.; Osman, I.; Stein, J.A. Analysis of recurrence patterns in acral versus nonacral melanoma: Should histologic subtype influence treatment guidelines? J. Natl. Compr. Cancer Netw. JNCCN 2014, 12, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Chow, H.Y.; Jubb, A.M.; Koch, J.N.; Jaffer, Z.M.; Stepanova, D.; Campbell, D.A.; Duron, S.G.; O’Farrell, M.; Cai, K.Q.; Klein-Szanto, A.J.; et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012, 72, 5966–5975. [Google Scholar] [CrossRef]
- Nheu, T.; He, H.; Hirokawa, Y.; Walker, F.; Wood, J.; Maruta, H. PAK is essential for RAS-induced upregulation of cyclin D1 during the G1 to S transition. Cell Cycle 2004, 3, 71–74. [Google Scholar] [CrossRef]
- Tang, Y.; Yu, J.; Field, J. Signals from the Ras, Rac, and Rho GTPases converge on the Pak protein kinase in Rat-1 fibroblasts. Mol. Cell. Biol. 1999, 19, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.G.; Chen, J.; Kirn, D.; McCormick, F.; Symons, M. An essential role for Rac in Ras transformation. Nature 1995, 374, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Tanda, E.T.; Dalmasso, B.; Pastorino, L.; Andreotti, V.; Bruno, W.; Boutros, A.; Spagnolo, F.; Ghiorzo, P. Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front. Mol. Biosci. 2020, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Lissanu Deribe, Y.; Shi, Y.; Rai, K.; Nezi, L.; Amin, S.B.; Wu, C.C.; Akdemir, K.C.; Mahdavi, M.; Peng, Q.; Chang, Q.E.; et al. Truncating PREX2 mutations activate its GEF activity and alter gene expression regulation in NRAS-mutant melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E1296–E1305. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., 3rd; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef]
- McDaniel, A.S.; Allen, J.D.; Park, S.J.; Jaffer, Z.M.; Michels, E.G.; Burgin, S.J.; Chen, S.; Bessler, W.K.; Hofmann, C.; Ingram, D.A.; et al. Pak1 regulates multiple c-Kit mediated Ras-MAPK gain-in-function phenotypes in Nf1+/- mast cells. Blood 2008, 112, 4646–4654. [Google Scholar] [CrossRef]
- Lodde, G.C.; Jansen, P.; Herbst, R.; Terheyden, P.; Utikal, J.; Pfohler, C.; Ulrich, J.; Kreuter, A.; Mohr, P.; Gutzmer, R.; et al. Characterisation and outcome of RAC1 mutated melanoma. Eur. J. Cancer 2023, 183, 1–10. [Google Scholar] [CrossRef]
- Vaque, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- McCarty, S.K.; Saji, M.; Zhang, X.; Knippler, C.M.; Kirschner, L.S.; Fernandez, S.; Ringel, M.D. BRAF activates and physically interacts with PAK to regulate cell motility. Endocr.-Relat. Cancer 2014, 21, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Serrone, L.; Zeuli, M.; Sega, F.M.; Cognetti, F. Dacarbazine-based chemotherapy for metastatic melanoma: Thirty-year experience overview. J. Exp. Clin. Cancer Res. CR 2000, 19, 21–34. [Google Scholar] [PubMed]
- Melia, B.M.; Abramson, D.H.; Albert, D.M.; Boldt, H.C.; Earle, J.D.; Hanson, W.F.; Montague, P.; Moy, C.S.; Schachat, A.P.; Simpson, E.R.; et al. Collaborative ocular melanoma study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years COMS report no. 16. Ophthalmology 2001, 108, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, A.; Cozzio, A.; Plasswilm, L.; Panje, C.M. Radiotherapy for lentigo maligna and lentigo maligna melanoma—A systematic review. Radiat. Oncol. 2020, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.R.; Mehnert, J.M. Mucosal Melanoma: Epidemiology, Biology and Treatment. Cancer Treat. Res. 2016, 167, 295–320. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.A.; Burmeister, B.H.; Ainslie, J.; Fisher, R.; Di Iulio, J.; Smithers, B.M.; Hong, A.; Shannon, K.; Scolyer, R.A.; Carruthers, S.; et al. Adjuvant lymph-node field radiotherapy versus observation only in patients with melanoma at high risk of further lymph-node field relapse after lymphadenectomy (ANZMTG 01.02/TROG 02.01): 6-year follow-up of a phase 3, randomised controlled trial. Lancet Oncol. 2015, 16, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, G.B.; Hong, A. Radiation therapy for advanced and metastatic melanoma. J. Surg. Oncol. 2014, 109, 370–375. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Tang, Y.; Liu, S.J.; Zeng, P.H.; Qu, L.; Jing, Q.C.; Yin, W.J. Radiation-induced liver disease: Beyond DNA damage. Cell Cycle 2023, 22, 506–526. [Google Scholar] [CrossRef] [PubMed]
- Roig, J.; Traugh, J.A. p21-activated protein kinase gamma-PAK is activated by ionizing radiation and other DNA-damaging agents. Similarities and differences to alpha-PAK. J. Biol. Chem. 1999, 274, 31119–31122. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Saldivar-Ceron, H.I.; Villamar-Cruz, O.; Perez-Plasencia, C.; Arias-Romero, L.E. p21 Activated kinase 1: Nuclear activity and its role during DNA damage repair. DNA Repair 2018, 65, 42–46. [Google Scholar] [CrossRef]
- Motwani, M.; Li, D.Q.; Horvath, A.; Kumar, R. Identification of novel gene targets and functions of p21-activated kinase 1 during DNA damage by gene expression profiling. PLoS ONE 2013, 8, e66585. [Google Scholar] [CrossRef]
- Ho, H.; Aruri, J.; Kapadia, R.; Mehr, H.; White, M.A.; Ganesan, A.K. RhoJ regulates melanoma chemoresistance by suppressing pathways that sense DNA damage. Cancer Res. 2012, 72, 5516–5528. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Bagheri-Yarmand, R.; Yang, Z.; Balasenthil, S.; Nguyen, D.; Sahin, A.A.; den Hollander, P.; Kumar, R. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell 2004, 5, 575–585. [Google Scholar] [CrossRef]
- Schurmann, A.; Mooney, A.F.; Sanders, L.C.; Sells, M.A.; Wang, H.G.; Reed, J.C.; Bokoch, G.M. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol. Cell. Biol. 2000, 20, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhuo, Y.; Guo, W.; Field, J. p21-activated Kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J. Biol. Chem. 2005, 280, 24698–24705. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Kumar, R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003, 535, 6–10. [Google Scholar] [CrossRef]
- Frost, J.A.; Swantek, J.L.; Stippec, S.; Yin, M.J.; Gaynor, R.; Cobb, M.H. Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J. Biol. Chem. 2000, 275, 19693–19699. [Google Scholar] [CrossRef]
- Kandel, E.S.; Lu, T.; Wan, Y.; Agarwal, M.K.; Jackson, M.W.; Stark, G.R. Mutagenesis by reversible promoter insertion to study the activation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2005, 102, 6425–6430. [Google Scholar] [CrossRef]
- Huang, C.; Radi, R.H.; Arbiser, J.L. Mitochondrial Metabolism in Melanoma. Cells 2021, 10, 3197. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef] [PubMed]
- Beeser, A.; Jaffer, Z.M.; Hofmann, C.; Chernoff, J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J. Biol. Chem. 2005, 280, 36609–36615. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.A.; Xu, S.; Hutchison, M.R.; Marcus, S.; Cobb, M.H. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell. Biol. 1996, 16, 3707–3713. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef] [PubMed]
- Eblen, S.T.; Slack, J.K.; Weber, M.J.; Catling, A.D. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol. Cell. Biol. 2002, 22, 6023–6033. [Google Scholar] [CrossRef] [PubMed]
- Broit, N.; Johansson, P.A.; Rodgers, C.B.; Walpole, S.T.; Newell, F.; Hayward, N.K.; Pritchard, A.L. Meta-Analysis and Systematic Review of the Genomics of Mucosal Melanoma. Mol. Cancer Res. 2021, 19, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Wróblewska, J.P.; Dias-Santagata, D.; Ustaszewski, A.; Wu, C.L.; Fujimoto, M.; Selim, M.A.; Biernat, W.; Ryś, J.; Marszalek, A.; Hoang, M.P. Prognostic Roles of BRAF, KIT, NRAS, IGF2R and SF3B1 Mutations in Mucosal Melanomas. Cells 2021, 10, 2216. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; Liszkay, G.; et al. COLUMBUS 5-Year Update: A Randomized, Open-Label, Phase III Trial of Encorafenib Plus Binimetinib Versus Vemurafenib or Encorafenib in Patients With BRAF V600-Mutant Melanoma. J. Clin. Oncol. 2022, 40, 4178–4188. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dreno, B.; Larkin, J.; Ribas, A.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. 5-Year Outcomes with Cobimetinib plus Vemurafenib in BRAFV600 Mutation-Positive Advanced Melanoma: Extended Follow-up of the coBRIM Study. Clin. Cancer Res. 2021, 27, 5225–5235. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Lebbe, C.; Dutriaux, C.; Lesimple, T.; Kruit, W.; Kerger, J.; Thomas, L.; Guillot, B.; Braud, F.; Garbe, C.; Grob, J.J.; et al. Pimasertib Versus Dacarbazine in Patients With Unresectable NRAS-Mutated Cutaneous Melanoma: Phase II, Randomized, Controlled Trial with Crossover. Cancers 2020, 12, 1727. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Patel, S.P.; Shoushtari, A.N.; Ambrosini, G.; Cremers, S.; Lee, S.; Franks, L.; Singh-Kandah, S.; Hernandez, S.; Sender, N.; et al. Intermittent MEK inhibition for the treatment of metastatic uveal melanoma. Front. Oncol. 2022, 12, 975643. [Google Scholar] [CrossRef] [PubMed]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef]
- Shao, Y.; Aplin, A.E. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010, 70, 6670–6681. [Google Scholar] [CrossRef]
- Kandel, E.S.; Hay, N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell. Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef] [PubMed]
- Babagana, M.; Kichina, J.V.; Slabodkin, H.; Johnson, S.; Maslov, A.; Brown, L.; Attwood, K.; Nikiforov, M.A.; Kandel, E.S. The role of polo-like kinase 3 in the response of BRAF-mutant cells to targeted anticancer therapies. Mol. Carcinog. 2020, 59, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Elion, E.A. Pheromone response, mating and cell biology. Curr. Opin. Microbiol. 2000, 3, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Vijai, J.; Kichina, J.V.; Byzova, T.; Kandel, E.S. The role of PAK-1 in activation of MAP kinase cascade and oncogenic transformation by Akt. Oncogene 2009, 28, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, H.; Chen, A.; Pittman, R.N.; Field, J. The Akt proto-oncogene links Ras to Pak and cell survival signals. J. Biol. Chem. 2000, 275, 9106–9109. [Google Scholar] [CrossRef]
- Singhal, R.; Kandel, E.S. The response to PAK1 inhibitor IPA3 distinguishes between cancer cells with mutations in BRAF and Ras oncogenes. Oncotarget 2012, 3, 700–708. [Google Scholar] [CrossRef]
- Lu, H.; Liu, S.; Zhang, G.; Bin, W.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef]
- Ruiz, R.; Jahid, S.; Harris, M.; Marzese, D.M.; Espitia, F.; Vasudeva, P.; Chen, C.F.; de Feraudy, S.; Wu, J.; Gillen, D.L.; et al. The RhoJ-BAD signaling network: An Achilles’ heel for BRAF mutant melanomas. PLoS Genet. 2017, 13, e1006913. [Google Scholar] [CrossRef] [PubMed]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Stejerean-Todoran, I.; Gimotty, P.A.; Watters, A.; Brafford, P.; Krepler, C.; Godok, T.; Li, H.; Bonilla Del Rio, Z.; Zieseniss, A.; Katschinski, D.M.; et al. A distinct pattern of growth and RAC1 signaling in melanoma brain metastasis cells. Neuro-Oncology 2023, 25, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Weber, J.S. Taming the wild-types: Targeting PAK1 in melanomas that lack BRAF mutations. J. Natl. Cancer Inst. 2013, 105, 591–592. [Google Scholar] [CrossRef]
- Verbik, D.J.; Murray, T.G.; Tran, J.M.; Ksander, B.R. Melanomas that develop within the eye inhibit lymphocyte proliferation. Int. J. Cancer 1997, 73, 470–478. [Google Scholar] [CrossRef]
- Babagana, M.; Brown, L.R.; Slabodkin, H.Z.; Kichina, J.V.; Kandel, E.S. Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT. Int. J. Mol. Sci. 2021, 22, 11367. [Google Scholar] [CrossRef] [PubMed]
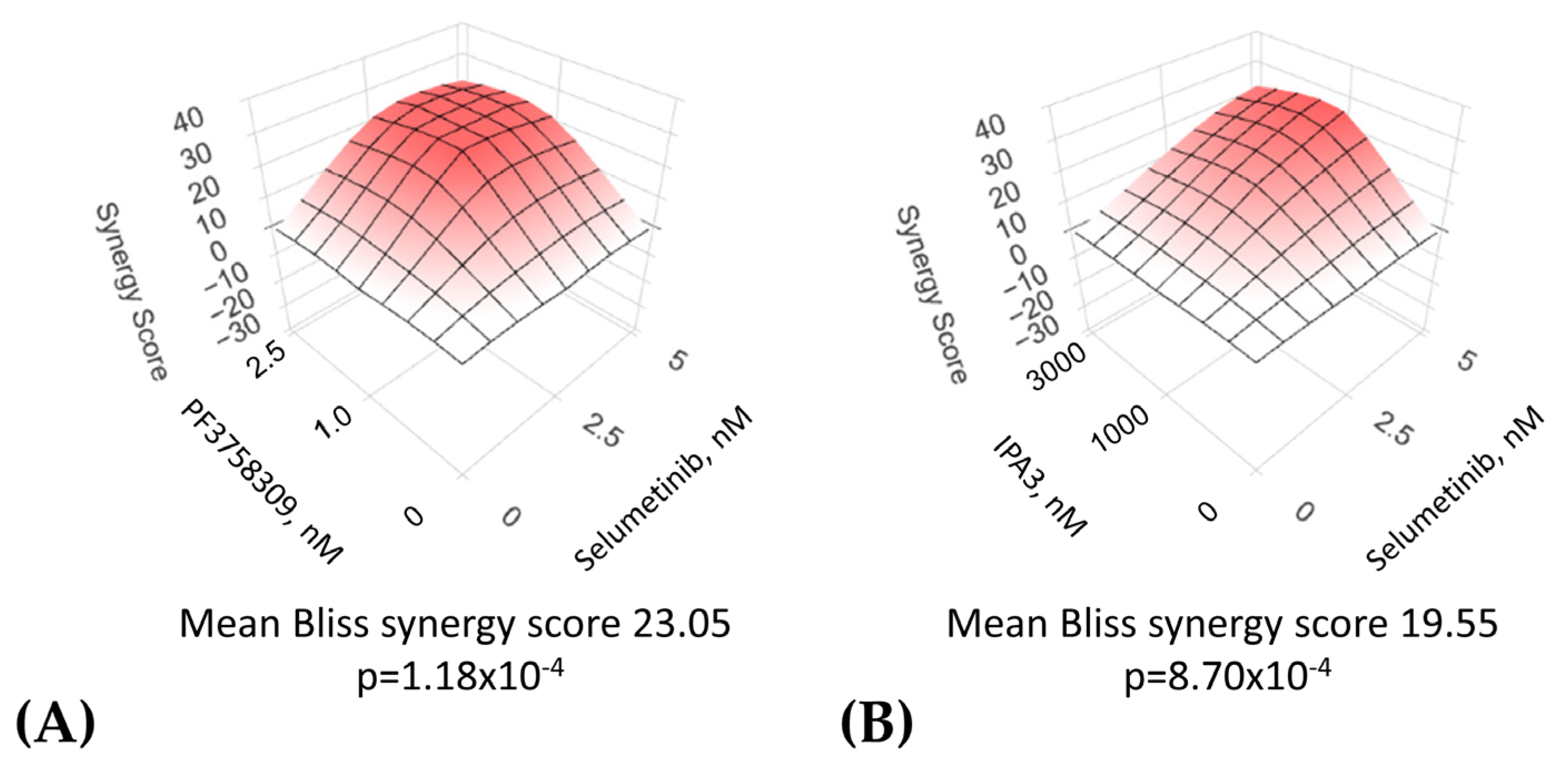
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Kissil, J.L.; Johnson, K.C.; Eckman, M.S.; Jacks, T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J. Biol. Chem. 2002, 277, 10394–10399. [Google Scholar] [CrossRef]
- Kissil, J.L.; Wilker, E.W.; Johnson, K.C.; Eckman, M.S.; Yaffe, M.B.; Jacks, T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol. Cell 2003, 12, 841–849. [Google Scholar] [CrossRef]
- Xiao, G.H.; Beeser, A.; Chernoff, J.; Testa, J.R. p21-activated kinase links Rac/Cdc42 signaling to merlin. J. Biol. Chem. 2002, 277, 883–886. [Google Scholar] [CrossRef]
- Sabra, H.; Brunner, M.; Mandati, V.; Wehrle-Haller, B.; Lallemand, D.; Ribba, A.S.; Chevalier, G.; Guardiola, P.; Block, M.R.; Bouvard, D. beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 2017, 292, 19179–19197. [Google Scholar] [CrossRef] [PubMed]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernández-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Piñeiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351. [Google Scholar] [CrossRef]
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.; Wang, K.; Yim, M.; Dumesny, C.J.; Sandrin, M.S.; Baldwin, G.S.; Nikfarjam, M.; He, H. Depletion of p21-activated kinase 1 up-regulates the immune system of APC(∆14/+) mice and inhibits intestinal tumorigenesis. BMC Cancer 2017, 17, 431. [Google Scholar] [CrossRef]
- Mimura, K.; Kua, L.F.; Shiraishi, K.; Kee Siang, L.; Shabbir, A.; Komachi, M.; Suzuki, Y.; Nakano, T.; Yong, W.P.; So, J.; et al. Inhibition of mitogen-activated protein kinase pathway can induce upregulation of human leukocyte antigen class I without PD-L1-upregulation in contrast to interferon-gamma treatment. Cancer Sci. 2014, 105, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Yang, Z.; Algazi, A.P.; Lomeli, S.H.; Wang, Y.; Othus, M.; Hong, A.; Wang, X.; Randolph, C.E.; et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell 2021, 39, 1375–1387.e1376. [Google Scholar] [CrossRef]
- Phadke, M.S.; Chen, Z.; Li, J.; Mohamed, E.; Davies, M.A.; Smalley, I.; Duckett, D.R.; Palve, V.; Czerniecki, B.J.; Forsyth, P.A.; et al. Targeted Therapy Given after Anti-PD-1 Leads to Prolonged Responses in Mouse Melanoma Models through Sustained Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Kennedy, S.A.; Jarboui, M.A.; Srihari, S.; Raso, C.; Bryan, K.; Dernayka, L.; Charitou, T.; Bernal-Llinares, M.; Herrera-Montavez, C.; Krstic, A.; et al. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS(G13D). Nat. Commun. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Vu, H.L.; Rosenbaum, S.; Purwin, T.J.; Davies, M.A.; Aplin, A.E. RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 2015, 28, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhan, Y.; Huynh, N.; Dumesny, C.; Wang, X.; Asadi, K.; Herrmann, D.; Timpson, P.; Yang, Y.; Walsh, K.; et al. Inhibition of PAK1 suppresses pancreatic cancer by stimulation of anti-tumour immunity through down-regulation of PD-L1. Cancer Lett. 2020, 472, 8–18. [Google Scholar] [CrossRef]
- Hahne, M.; Rimoldi, D.; Schroter, M.; Romero, P.; Schreier, M.; French, L.E.; Schneider, P.; Bornand, T.; Fontana, A.; Lienard, D.; et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: Implications for tumor immune escape. Science 1996, 274, 1363–1366. [Google Scholar] [CrossRef]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Chappell, D.B.; Zaks, T.Z.; Rosenberg, S.A.; Restifo, N.P. Human melanoma cells do not express Fas (Apo-1/CD95) ligand. Cancer Res. 1999, 59, 59–62. [Google Scholar]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Dumont, C.; Cruz, A.C.; Muppidi, J.R.; Gomez, T.S.; Billadeau, D.D.; Tybulewicz, V.L.; Siegel, R.M. Cutting edge: Rac GTPases sensitize activated T cells to die via Fas. J. Immunol. 2007, 179, 6384–6388. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Mohan, A.A.; Dennison, L.; Vithayathil, T.; Ruggieri, A.; Lesinski, G.B.; Armstrong, T.D.; Azad, N.S.; Jaffee, E.M. MEK inhibition suppresses B regulatory cells and augments anti-tumor immunity. PLoS ONE 2019, 14, e0224600. [Google Scholar] [CrossRef] [PubMed]
- Semenova, G.; Chernoff, J. Targeting PAK1. Biochem. Soc. Trans. 2017, 45, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Jaffer, Z.M.; Park, S.J.; Burgin, S.; Hofmann, C.; Sells, M.A.; Chen, S.; Derr-Yellin, E.; Michels, E.G.; McDaniel, A.; et al. p21-activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 2009, 113, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Lei, M.; Wang, X.; Solaro, R.J. Novel roles of PAK1 in the heart. Cell. Logist. 2012, 2, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zynda, E.R.; Maloy, M.H.; Kandel, E.S. The role of PAK1 in the sensitivity of kidney epithelial cells to ischemia-like conditions. Cell Cycle 2019, 18, 596–604. [Google Scholar] [CrossRef]
- Marlin, J.W.; Chang, Y.W.; Ober, M.; Handy, A.; Xu, W.; Jakobi, R. Functional PAK-2 knockout and replacement with a caspase cleavage-deficient mutant in mice reveals differential requirements of full-length PAK-2 and caspase-activated PAK-2p34. Mamm. Genome 2011, 22, 306–317. [Google Scholar] [CrossRef]
- Hofmann, C.; Shepelev, M.; Chernoff, J. The genetics of Pak. J. Cell Sci. 2004, 117, 4343–4354. [Google Scholar] [CrossRef]
- Meng, J.; Meng, Y.; Hanna, A.; Janus, C.; Jia, Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. 2005, 25, 6641–6650. [Google Scholar] [CrossRef]
- Chow, H.Y.; Karchugina, S.; Groendyke, B.J.; Toenjes, S.; Hatcher, J.; Donovan, K.A.; Fischer, E.S.; Abalakov, G.; Faezov, B.; Dunbrack, R.; et al. Development and Utility of a PAK1-Selective Degrader. J. Med. Chem. 2022, 65, 15627–15641. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.S.; Amiri, P.; Bellamacina, C.; Bellance, M.H.; Breitenstein, W.; Daniel, D.; Denay, R.; Fabbro, D.; Fernandez, C.; Galuba, I.; et al. Optimization of a Dibenzodiazepine Hit to a Potent and Selective Allosteric PAK1 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Bricelj, A.; Steinebach, C.; Kuchta, R.; Gütschow, M.; Sosič, I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 2021, 9, 707317. [Google Scholar] [CrossRef] [PubMed]
- Thompson, Z.J.; Teer, J.K.; Li, J.; Chen, Z.; Welsh, E.A.; Zhang, Y.; Ayoubi, N.; Eroglu, Z.; Tan, A.C.; Smalley, K.S.M.; et al. Drepmel-A Multi-Omics Melanoma Drug Repurposing Resource for Prioritizing Drug Combinations and Understanding Tumor Microenvironment. Cells 2022, 11, 2894. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; Crawford, J.J.; Hoeflich, K.P.; Wang, W. Inhibitors of p21-activated kinases (PAKs). J. Med. Chem. 2015, 58, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Missaoui, W.N.; Cummings, B.S.; Somanath, P.R. Liposome-mediated delivery of the p21 activated kinase-1 (PAK-1) inhibitor IPA-3 limits prostate tumor growth in vivo. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Najahi-Missaoui, W.; Quach, N.D.; Somanath, P.R.; Cummings, B.S. Liposomes Targeting P21 Activated Kinase-1 (PAK-1) and Selective for Secretory Phospholipase A(2) (sPLA(2)) Decrease Cell Viability and Induce Apoptosis in Metastatic Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9396. [Google Scholar] [CrossRef]
- Bailly, C.; Beignet, J.; Loirand, G.; Sauzeau, V. Rac1 as a therapeutic anticancer target: Promises and limitations. Biochem. Pharmacol. 2022, 203, 115180. [Google Scholar] [CrossRef]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef]
- Bianchi-Smiraglia, A.; Wolff, D.W.; Marston, D.J.; Deng, Z.; Han, Z.; Moparthy, S.; Wombacher, R.M.; Mussell, A.L.; Shen, S.; Chen, J.; et al. Regulation of local GTP availability controls RAC1 activity and cell invasion. Nat. Commun. 2021, 12, 6091. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef]
- Barnaba, N.; LaRocque, J.R. Targeting cell cycle regulation via the G2-M checkpoint for synthetic lethality in melanoma. Cell Cycle 2021, 20, 1041–1051. [Google Scholar] [CrossRef]
- Maresca, L.; Stecca, B.; Carrassa, L. Novel Therapeutic Approaches with DNA Damage Response Inhibitors for Melanoma Treatment. Cells 2022, 11, 1466. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kichina, J.V.; Maslov, A.; Kandel, E.S. PAK1 and Therapy Resistance in Melanoma. Cells 2023, 12, 2373. https://doi.org/10.3390/cells12192373
Kichina JV, Maslov A, Kandel ES. PAK1 and Therapy Resistance in Melanoma. Cells. 2023; 12(19):2373. https://doi.org/10.3390/cells12192373
Chicago/Turabian StyleKichina, Julia V., Alexei Maslov, and Eugene S. Kandel. 2023. "PAK1 and Therapy Resistance in Melanoma" Cells 12, no. 19: 2373. https://doi.org/10.3390/cells12192373
APA StyleKichina, J. V., Maslov, A., & Kandel, E. S. (2023). PAK1 and Therapy Resistance in Melanoma. Cells, 12(19), 2373. https://doi.org/10.3390/cells12192373