
Fractalkine/CX3CR1 in Dilated Cardiomyopathy: A Potential Future Target for Immunomodulatory Therapy?
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Phenotypic Clustering of Dilated Cardiomyopathy Patients
- PG1 included 331 patients with mild systolic dysfunction
- (Mean ejection fraction: 43% ± 9%),
- PG2 included 83 patients with auto-immune disease background
- PG3 included 165 patients with cardiac arrhythmias (mainly atrial fibrillation and ventricular tachycardias), including patients with genetic causes (Familial cardiomyopathy)
- PG4 included 216 patients with severe systolic dysfunction (Mean ejection fraction: 23% ± 8%)
3. Aetiologies of Dilated Cardiomyopathy
3.1. Familial Cardiomyopathy
3.2. Autoimmune Myocarditis
3.3. Post-Viral Myocarditis
3.4. Immune Checkpoint Inhibitor-Related Myocarditis
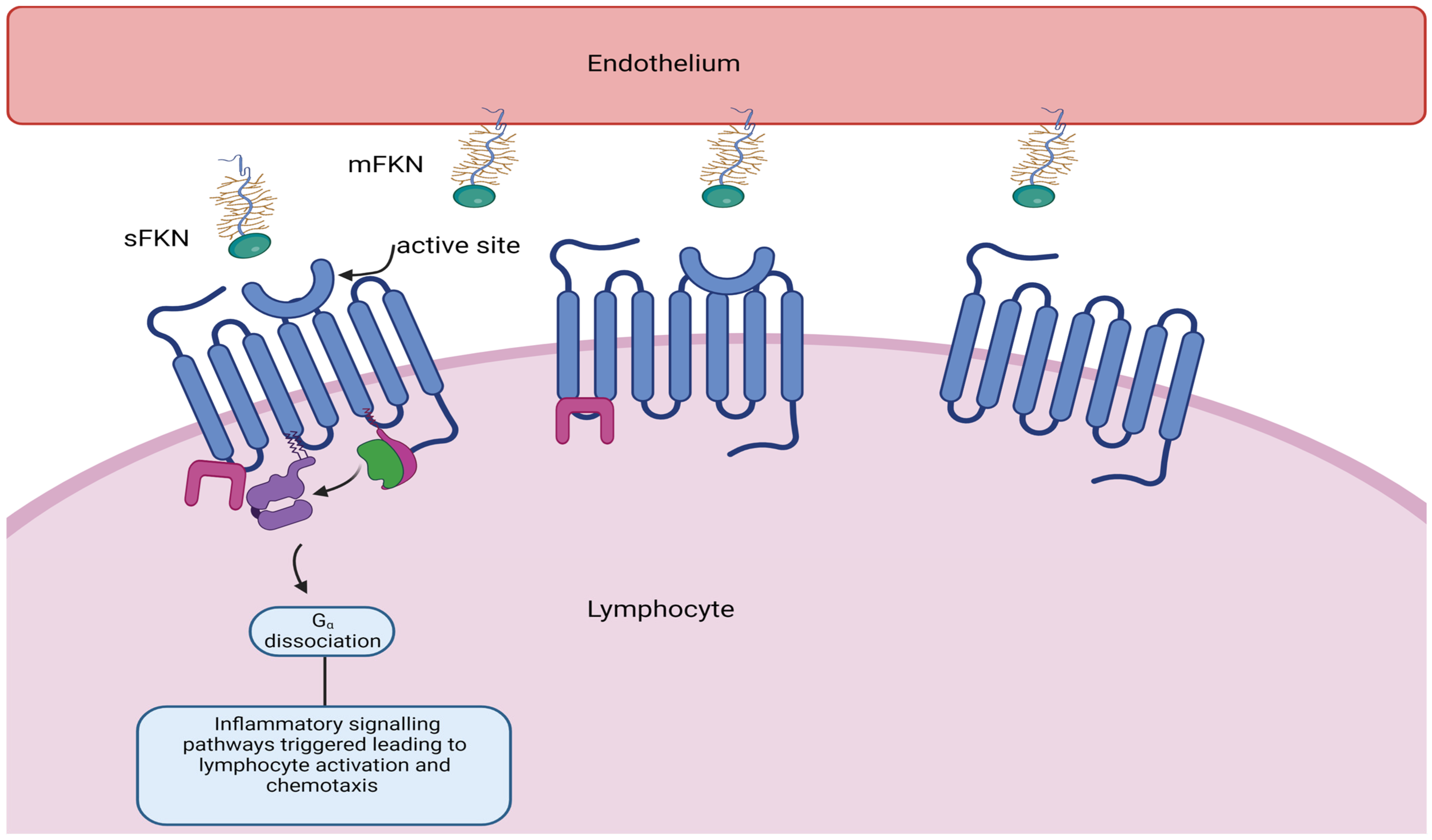
4. Role of Fractalkine Signalling in Cardiovascular Disease
4.1. Fractalkine (CX3CL1) and Its Receptor CX3CR1 in Atherogenesis
4.2. CX3CR1 and Myocardial Infarction
4.3. Fractalkine Signalling in Heart Failure
5. Link between CX3CR1 and Cytomegalovirus
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Helio, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- Kuhl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef]
- Kuhl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef]
- Nakayama, T.; Sugano, Y.; Yokokawa, T.; Nagai, T.; Matsuyama, T.A.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Ohte, N.; et al. Clinical impact of the presence of macrophages in endomyocardial biopsies of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 490–498. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Merlo, M.; Dominguez, F.; Wang, P.; Henkens, M.; Adriaens, M.E.; Hazebroek, M.R.; Mase, M.; Escobar, L.E.; Cobas-Paz, R.; et al. Phenotypic clustering of dilated cardiomyopathy patients highlights important pathophysiological differences. Eur. Heart J. 2021, 42, 162–174. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Comarmond, C.; Cacoub, P. Myocarditis in auto-immune or auto-inflammatory diseases. Autoimmun. Rev. 2017, 16, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Merken, J.; Hazebroek, M.; Van Paassen, P.; Verdonschot, J.; Van Empel, V.; Knackstedt, C.; Abdul Hamid, M.; Seiler, M.; Kolb, J.; Hoermann, P.; et al. Immunosuppressive Therapy Improves Both Short- and Long-Term Prognosis in Patients with Virus-Negative Nonfulminant Inflammatory Cardiomyopathy. Circ. Heart Fail. 2018, 11, e004228. [Google Scholar] [CrossRef]
- Scheinecker, C.; Goschl, L.; Bonelli, M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J. Autoimmun. 2020, 110, 102376. [Google Scholar] [CrossRef] [PubMed]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Olejniczak, M.; Schwartz, M.; Webber, E.; Shaffer, A.; Perry, T.E. Viral Myocarditis-Incidence, Diagnosis and Management. J. Cardiothorac. Vasc. Anesth. 2020, 34, 1591–1601. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis–diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef]
- Han, L.N.; He, S.; Wang, Y.T.; Yang, L.M.; Liu, S.Y.; Zhang, T. Advances in monoclonal antibody application in myocarditis. J. Zhejiang Univ. Sci. B 2013, 14, 676–687. [Google Scholar] [CrossRef]
- Jensen, L.D.; Marchant, D.J. Emerging pharmacologic targets and treatments for myocarditis. Pharmacol. Ther. 2016, 161, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Curnock, A.P.; Bossi, G.; Kumaran, J.; Bawden, L.J.; Figueiredo, R.; Tawar, R.; Wiseman, K.; Henderson, E.; Hoong, S.J.; Gonzalez, V.; et al. Cell-targeted PD-1 agonists that mimic PD-L1 are potent T cell inhibitors. JCI Insight 2021, 6, e152468. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Zafar, A.; Zubiri, L.; Zlotoff, D.A.; Alvi, R.M.; Lee, C.; Hartmann, S.; Gilman, H.K.; Villani, A.C.; Nohria, A.; et al. Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio with Immune Checkpoint Inhibitor-Associated Myocarditis. J. Am. Heart Assoc. 2020, 9, e018306. [Google Scholar] [CrossRef]
- Zhang, L.; Awadalla, M.; Mahmood, S.S.; Nohria, A.; Hassan, M.Z.O.; Thuny, F.; Zlotoff, D.A.; Murphy, S.P.; Stone, J.R.; Golden, D.L.A.; et al. Cardiovascular magnetic resonance in immune checkpoint inhibitor-associated myocarditis. Eur. Heart J. 2020, 41, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, S.; Neilan, T.G. Immune Checkpoint Inhibitor-Associated Myocarditis. Oncologist 2018, 23, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Palaskas, N.; Lopez-Mattei, J.; Durand, J.B.; Iliescu, C.; Deswal, A. Immune Checkpoint Inhibitor Myocarditis: Pathophysiological Characteristics, Diagnosis, and Treatment. J. Am. Heart Assoc. 2020, 9, e013757. [Google Scholar] [CrossRef]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef] [PubMed]
- Goda, S.; Imai, T.; Yoshie, O.; Yoneda, O.; Inoue, H.; Nagano, Y.; Okazaki, T.; Imai, H.; Bloom, E.T.; Domae, N.; et al. CX3C-chemokine, fractalkine-enhanced adhesion of THP-1 cells to endothelial cells through integrin-dependent and -independent mechanisms. J. Immunol. 2000, 164, 4313–4320. [Google Scholar] [CrossRef]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef]
- Kanazawa, N.; Nakamura, T.; Tashiro, K.; Muramatsu, M.; Morita, K.; Yoneda, K.; Inaba, K.; Imamura, S.; Honjo, T. Fractalkine and macrophage-derived chemokine: T cell-attracting chemokines expressed in T cell area dendritic cells. Eur. J. Immunol. 1999, 29, 1925–1932. [Google Scholar] [CrossRef]
- Patel, A.; Jagadesham, V.P.; Porter, K.E.; Scott, D.J.; Carding, S.R. Characterisation of fractalkine/CX3CL1 and fractalkine receptor (CX3CR1) expression in abdominal aortic aneurysm disease. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 20–27. [Google Scholar] [CrossRef]
- Nishimura, M.; Umehara, H.; Nakayama, T.; Yoneda, O.; Hieshima, K.; Kakizaki, M.; Dohmae, N.; Yoshie, O.; Imai, T. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme B+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J. Immunol. 2002, 168, 6173–6180. [Google Scholar] [CrossRef]
- Yoneda, O.; Imai, T.; Goda, S.; Inoue, H.; Yamauchi, A.; Okazaki, T.; Imai, H.; Yoshie, O.; Bloom, E.T.; Domae, N.; et al. Fractalkine-mediated endothelial cell injury by NK cells. J. Immunol. 2000, 164, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Husberg, C.; Nygard, S.; Finsen, A.V.; Damas, J.K.; Frigessi, A.; Oie, E.; Waehre, A.; Gullestad, L.; Aukrust, P.; Yndestad, A.; et al. Cytokine expression profiling of the myocardium reveals a role for CX3CL1 (fractalkine) in heart failure. J. Mol. Cell Cardiol. 2008, 45, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Combadière, C.; Potteaux, S.; Gao, J.L.; Esposito, B.; Casanova, S.; Lee, E.J.; Debré, P.; Tedgui, A.; Murphy, P.M.; Mallat, Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 2003, 107, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, P.; Haskell, C.A.; Charo, I.F. Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J. Clin. Investig. 2003, 111, 333–340. [Google Scholar] [CrossRef]
- Apostolakis, S.; Amanatidou, V.; Papadakis, E.G.; Spandidos, D.A. Genetic diversity of CX3CR1 gene and coronary artery disease: New insights through a meta-analysis. Atherosclerosis 2009, 207, 8–15. [Google Scholar] [CrossRef]
- Apostolakis, S.; Baritaki, S.; Kochiadakis, G.E.; Igoumenidis, N.E.; Panutsopulos, D.; Spandidos, D.A. Effects of polymorphisms in chemokine ligands and receptors on susceptibility to coronary artery disease. Thromb. Res. 2007, 119, 63–71. [Google Scholar] [CrossRef]
- Chan, C.C.; Tuo, J.; Bojanowski, C.M.; Csaky, K.G.; Green, W.R. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol. Histopathol. 2005, 20, 857–863. [Google Scholar] [CrossRef]
- Ghilardi, G.; Biondi, M.L.; Turri, O.; Guagnellini, E.; Scorza, R. Internal carotid artery occlusive disease and polymorphisms of fractalkine receptor CX3CR1: A genetic risk factor. Stroke 2004, 35, 1276–1279. [Google Scholar] [CrossRef]
- Hattori, H.; Ito, D.; Tanahashi, N.; Murata, M.; Saito, I.; Watanabe, K.; Suzuki, N. T280M and V249I polymorphisms of fractalkine receptor CX3CR1 and ischemic cerebrovascular disease. Neurosci. Lett. 2005, 374, 132–135. [Google Scholar] [CrossRef]
- Kimouli, M.; Miyakis, S.; Georgakopoulos, P.; Neofytou, E.; Achimastos, A.D.; Spandidos, D.A. Polymorphisms of fractalkine receptor CX3CR1 gene in patients with symptomatic and asymptomatic carotid artery stenosis. J. Atheroscler. Thromb. 2009, 16, 604–610. [Google Scholar] [CrossRef]
- McDermott, D.H.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Merrell, M.N.; Epstein, N.; Quyyumi, A.A.; Murphy, P.M. Association between polymorphism in the chemokine receptor CX3CR1 and coronary vascular endothelial dysfunction and atherosclerosis. Circ. Res. 2001, 89, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Garlaschelli, K.; Ongari, M.; Raselli, S.; Grigore, L.; Catapano, A.L. Effects of fractalkine receptor variants on common carotid artery intima-media thickness. Stroke 2006, 37, 1558–1561. [Google Scholar] [CrossRef]
- Wu, J.; Yin, R.X.; Lin, Q.Z.; Guo, T.; Shi, G.Y.; Sun, J.Q.; Shen, S.W.; Li, Q. Two polymorphisms in the Fractalkine receptor CX3CR1 gene influence the development of atherosclerosis: A meta-analysis. Dis. Markers 2014, 2014, 913678. [Google Scholar] [CrossRef] [PubMed]
- Boag, S.E.; Das, R.; Shmeleva, E.V.; Bagnall, A.; Egred, M.; Howard, N.; Bennaceur, K.; Zaman, A.; Keavney, B.; Spyridopoulos, I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J. Clin. Investig. 2015, 125, 3063–3076. [Google Scholar] [CrossRef] [PubMed]
- Umehara, H.; Bloom, E.T.; Okazaki, T.; Nagano, Y.; Yoshie, O.; Imai, T. Fractalkine in vascular biology: From basic research to clinical disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 34–40. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Martin-Ruiz, C.; Hilkens, C.; Yadegarfar, M.E.; Isaacs, J.; Jagger, C.; Kirkwood, T.; von Zglinicki, T. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: Results from the Newcastle 85+ study. Aging Cell 2016, 15, 389–392. [Google Scholar] [CrossRef]
- Strindhall, J.; Skog, M.; Ernerudh, J.; Bengner, M.; Lofgren, S.; Matussek, A.; Nilsson, B.O.; Wikby, A. The inverted CD4/CD8 ratio and associated parameters in 66-year-old individuals: The Swedish HEXA immune study. Age 2013, 35, 985–991. [Google Scholar] [CrossRef]
- Pachnio, A.; Ciaurriz, M.; Begum, J.; Lal, N.; Zuo, J.; Beggs, A.; Moss, P. Cytomegalovirus Infection Leads to Development of High Frequencies of Cytotoxic Virus-Specific CD4+ T Cells Targeted to Vascular Endothelium. PLoS Pathog. 2016, 12, e1005832. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Vossen, M.T.; Gent, M.R.; Davin, J.C.; Roos, M.T.; Wertheim-van Dillen, P.M.; Weel, J.F.; Baars, P.A.; van Lier, R.A. Frequencies of circulating cytolytic, CD45RA+CD27−, CD8+ T lymphocytes depend on infection with CMV. J. Immunol. 2003, 170, 4342–4348. [Google Scholar] [CrossRef]
- Pekalski, M.; Jenkinson, S.E.; Willet, J.D.; Poyner, E.F.; Alhamidi, A.H.; Robertson, H.; Ali, S.; Kirby, J.A. Renal allograft rejection: Examination of delayed differentiation of Treg and Th17 effector T cells. Immunobiology 2013, 218, 303–310. [Google Scholar] [CrossRef]
- Willet, J.D.; Pichitsiri, W.; Jenkinson, S.E.; Brain, J.G.; Wood, K.; Alhasan, A.A.; Spielhofer, J.; Robertson, H.; Ali, S.; Kirby, J.A. Kidney transplantation: Analysis of the expression and T cell-mediated activation of latent TGF-beta. J. Leukoc. Biol. 2013, 93, 471–478. [Google Scholar] [CrossRef] [PubMed]
- de Dios, E.; Rios-Navarro, C.; Perez-Sole, N.; Gavara, J.; Marcos-Garces, V.; Rodriguez, E.; Carratala, A.; Forner, M.J.; Navarro, J.; Blasco, M.L.; et al. Similar Clinical Course and Significance of Circulating Innate and Adaptive Immune Cell Counts in STEMI and COVID-19. J. Clin. Med. 2020, 9, 3484. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Koller, L.; Hohensinner, P.J.; Rychli, K.; Zorn, G.; Goliasch, G.; Berger, R.; Mortl, D.; Maurer, G.; Huber, K.; et al. Fractalkine is an independent predictor of mortality in patients with advanced heart failure. Thromb. Haemost. 2012, 108, 1220–1227. [Google Scholar] [CrossRef]
- Weltevrede, M.; Eilers, R.; de Melker, H.E.; van Baarle, D. Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: A systematic review. Exp. Gerontol. 2016, 77, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Waller, E.C.; Day, E.; Sissons, J.G.; Wills, M.R. Dynamics of T cell memory in human cytomegalovirus infection. Med. Microbiol. Immunol. 2008, 197, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Pourgheysari, B.; Khan, N.; Best, D.; Bruton, R.; Nayak, L.; Moss, P.A. The cytomegalovirus-specific CD4+ T-cell response expands with age and markedly alters the CD4+ T-cell repertoire. J. Virol. 2007, 81, 7759–7765. [Google Scholar] [CrossRef] [PubMed]
- Munks, M.W.; Cho, K.S.; Pinto, A.K.; Sierro, S.; Klenerman, P.; Hill, A.B. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol. 2006, 177, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Hislop, A.; Gudgeon, N.; Cobbold, M.; Khanna, R.; Nayak, L.; Rickinson, A.B.; Moss, P.A. Herpesvirus-specific CD8 T cell immunity in old age: Cytomegalovirus impairs the response to a coresident EBV infection. J. Immunol. 2004, 173, 7481–7489. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Successful and Maladaptive T Cell Aging. Immunity 2017, 46, 364–378. [Google Scholar] [CrossRef]
- Bolovan-Fritts, C.A.; Trout, R.N.; Spector, S.A. High T-cell response to human cytomegalovirus induces chemokine-mediated endothelial cell damage. Blood 2007, 110, 1857–1863. [Google Scholar] [CrossRef]
- Bolovan-Fritts, C.A.; Trout, R.N.; Spector, S.A. Human cytomegalovirus-specific CD4+-T-cell cytokine response induces fractalkine in endothelial cells. J. Virol. 2004, 78, 13173–13181. [Google Scholar] [CrossRef]
- Bolovan-Fritts, C.A.; Spector, S.A. Endothelial damage from cytomegalovirus-specific host immune response can be prevented by targeted disruption of fractalkine-CX3CR1 interaction. Blood 2008, 111, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Hertoghs, K.M.; Moerland, P.D.; van Stijn, A.; Remmerswaal, E.B.; Yong, S.L.; van de Berg, P.J.; van Ham, S.M.; Baas, F.; ten Berge, I.J.; van Lier, R.A. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J. Clin. Investig. 2010, 120, 4077–4090. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.N.; An, L.; Zhan, P.; Chen, X.H. Cytomegalovirus infection and coronary heart disease risk: A meta-analysis. Mol. Biol. Rep. 2012, 39, 6537–6546. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Sun, Y.; Pinto, C.; Camus, S.; Duval, V.; Alayrac, P.; Zlatanova, I.; Loyer, X.; Vilar, J.; Lemitre, M.; Levoye, A.; et al. Splenic Marginal Zone B Lymphocytes Regulate Cardiac Remodeling After Acute Myocardial Infarction in Mice. J. Am. Coll. Cardiol. 2022, 79, 632–647. [Google Scholar] [CrossRef]
- Stanier, P.; Taylor, D.L.; Kitchen, A.D.; Wales, N.; Tryhorn, Y.; Tyms, A.S. Persistence of cytomegalovirus in mononuclear cells in peripheral blood from blood donors. BMJ 1989, 299, 897–898. [Google Scholar] [CrossRef]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef]
- Siscovick, D.S.; Schwartz, S.M.; Corey, L.; Grayston, J.T.; Ashley, R.; Wang, S.P.; Psaty, B.M.; Tracy, R.P.; Kuller, L.H.; Kronmal, R.A. Chlamydia pneumoniae, herpes simplex virus type 1, and cytomegalovirus and incident myocardial infarction and coronary heart disease death in older adults: The Cardiovascular Health Study. Circulation 2000, 102, 2335–2340. [Google Scholar] [CrossRef]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies Up to 2016. J. Am. Heart Assoc. 2017, 6, e005025. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Auvynet, C.; Poupel, L.; Vilar, J.; Dumeau, E.; Richart, A.; Recalde, A.; Zouggari, Y.; Yin, K.Y.; Bruneval, P.; et al. The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abesser, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Shmeleva, E.V.; Boag, S.E.; Fiser, K.; Bagnall, A.; Murali, S.; Dimmick, I.; Pircher, H.; Martin-Ruiz, C.; Egred, M.; et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients. Circ. Res. 2015, 116, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulos, I.; Hoffmann, J.; Aicher, A.; Brummendorf, T.H.; Doerr, H.W.; Zeiher, A.M.; Dimmeler, S. Accelerated telomere shortening in leukocyte subpopulations of patients with coronary heart disease: Role of cytomegalovirus seropositivity. Circulation 2009, 120, 1364–1372. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Causes | LMNA44, MYH6, MYH7, MYBPC3, TNNT2, TTN46, RBM20, SCN5A, BAG3, PSEN3 | |
|---|---|---|
| Infections | Viruses | Adenovirus spp., Coronavirus spp., Coxsackievirus spp. (groups A and B), Cytomegalovirus spp., Dengue virus, Echovirus spp., Epstein-Barr virus, Hepatitis B virus, Hepatitis C virus, Herpes Simplex Virus, Human Herpesvirus 6, HIV, Influenza A and Influenza B viruses, Mumps rubulavirus, parvovirus (B19), poliovirus, Rabies virus, Respiratory syncytial virus, Rubella virus, Measles virus, and Varicella- zoster virus |
| Bacteria | ß-haemolytic streptococci, Borrelia burgdorferi, Brucella spp., Campylobacter jejuni, Chlamydia spp., Clostridium spp., Corynebacterium diphtheriae, Neisseria spp., Haemophilus influenza, Legionella pneumophila, Listeria monocytogenes, Mycoplasma pneumonia, Neisseria meningitidis, Salmonella (Berta and Typhi), Streptococcus pneumonia, Staphylococcus spp., and Treponema pallidum | |
| Protozoa | Entamoeba histolytica, Leishmania spp., Plasmodium vivax, Plasmodium falciparum, Toxoplasma gondii, and Trypanosoma cruzi | |
| Helminths | Taenia spp., Echinococcus spp., Schistosoma spp., Toxocara spp., and Trichinella spp. | |
| Fungi | Actinomyces spp., Aspergilus spp., Coccidioides immitis, and Cryptococcus neoformans | |
| Autoimmunity | Systemic sclerosis, rheumatoid arthritis, systemic lupus erythematosus, dermatomyositis, sarcoidosis, Dressler syndrome, post-cardiotomy syndrome, post-infectious autoimmune disease, and post-radiation autoimmune disease | |
| Toxin exposure | Alcohol, amphetamines, anthracyclines, cannabis, catecholamines, cocaine, 5-fluorouracil, lithium, heavy metals (cobalt, lead, and mercury), and carbon monoxide | |
| Metabolic or Endocrine | Cushing disease, hypothyroidism, hyperthyroidism, phaeochromocytoma, chronic hypocalcaemia, hypophosphataemia, and inborn errors of metabolism such as mitochondrial diseases and nutritional deficiency (carnitine, thiamine, and selenium) | |
| Pregnancy | Peripartum cardiomyopathy | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeyalan, V.; Austin, D.; Loh, S.X.; Wangsaputra, V.K.; Spyridopoulos, I. Fractalkine/CX3CR1 in Dilated Cardiomyopathy: A Potential Future Target for Immunomodulatory Therapy? Cells 2023, 12, 2377. https://doi.org/10.3390/cells12192377
Jeyalan V, Austin D, Loh SX, Wangsaputra VK, Spyridopoulos I. Fractalkine/CX3CR1 in Dilated Cardiomyopathy: A Potential Future Target for Immunomodulatory Therapy? Cells. 2023; 12(19):2377. https://doi.org/10.3390/cells12192377
Chicago/Turabian StyleJeyalan, Visvesh, David Austin, Shu Xian Loh, Vincent Kharisma Wangsaputra, and Ioakim Spyridopoulos. 2023. "Fractalkine/CX3CR1 in Dilated Cardiomyopathy: A Potential Future Target for Immunomodulatory Therapy?" Cells 12, no. 19: 2377. https://doi.org/10.3390/cells12192377