
Current Evidence Supporting the Role of Immune Response in ATTRv Amyloidosis

 , , ,
, , ,  and
and
Abstract
:1. Introduction
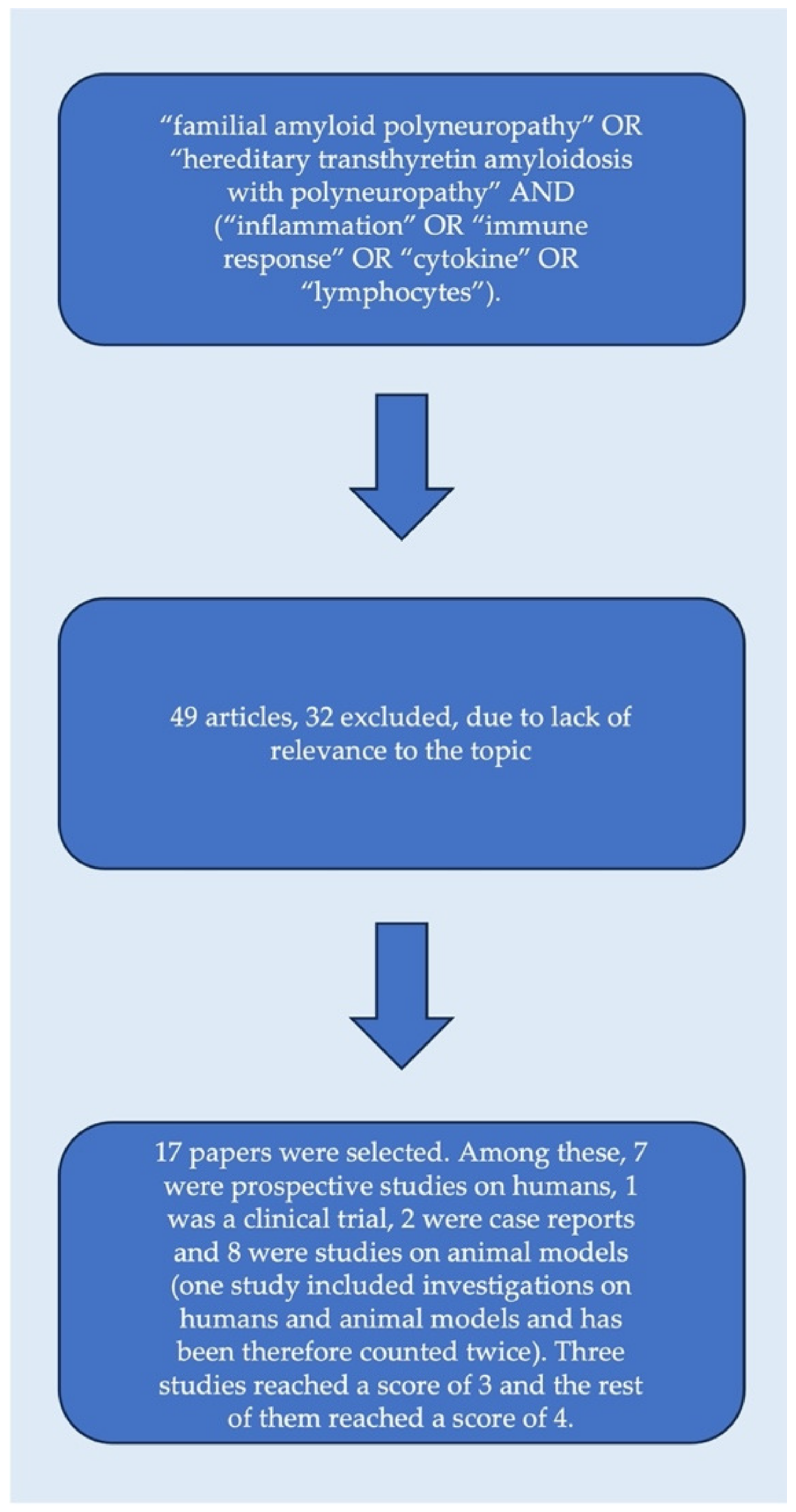
2. Materials and Methods
3. Hereditary Transthyretin Amyloidosis with Polyneuropathy: Current Pathogenetic View
4. Evidence of Immune Response in ATTRv Amyloidosis
4.1. Human Studies: State of the Field

{kind=link}
{kind=link}
| Reference | Methods and Techniques | Main Findings |
|---|---|---|
| Sousa et al., 2001 [52] | Analysis of nerve biopsy samples from patients by semiquantitative immunohistology and in situ hybridization | Increased levels of RAGE beginning at the earliest stages of the disease; upregulation of TNF-α, IL1- β, and iNOS in a distribution overlapping RAGE expression. |
| Matsunaga et al., 2002 [53] | IHC and sequential IF staining | RAGE and AGE have a distribution strongly correlated to that of amyloid deposits. However, no correlation was detected between NF-κB, apoptotic marker, and amyloid deposits. |
| Azevedo et al., 2019 [7] | ELISA | Increased serum levels of TNF-α, IL-1β, IL-8, IL-33, IFN-β and IL-10, and decreased levels of IL-12 in ATTRv patients. |
| Luigetti M et al., 2022 [8] | Luminex XMAP multiplexing technology | Increased serum levels of IFN-alpha and IFN-gamma, and decreased serum levels of IL-7 in ATTRv patients. |
| Suenaga et al., 2017 [6] | ELISA, cell culture, and Bio-Plex pro cytokine assay kit | IL-6 serum concentration was elevated in FAP carriers. In native TTR culture conditions, IL-6 increased in CD14 + monocytes in the presence of V30M-mutated TTR, compared with wild-type TTR, in a TTR-dose-dependent manner. IL-6 concentration increased in CD4 + T cells and CD8 + T cells in a TTR-dose-dependent manner. IL-1β, TNF-α, and IL-10 increased in a TTR-dose-dependent manner in CD14 + monocytes. |
| Kurian et al., 2016 [76] | Microarray technology and Luminex bead assays | Downregulation of eIF2 pathway in all symptomatic subjects, as well as primary immunodeficiency signaling, and purine nucleotide biosynthesis. Signaling networks for FCγ, TREM1, NK cells, IL3, IL15, and IL22 were all upregulated in FAP patients. Symptomatic females showed a downregulation of eIF2, primary immunodeficiency, T-helper cell differentiation, and iCOS signaling pathways. In symptomatic males, 29 significant canonical pathways linked to immunity, including Fcγ receptor, NK cell, Toll-like receptor, B-cell receptor, leukocyte etravasation, and IL-12 signaling, were all upregulated. There was a trend towards the normalization of all these altered gene expressions in patients treated with tafamidis. |
| Moreira et al., 2023 [67] | Real-time PCR, cell culture | Plasma levels of S100A8 protein were lower in ATTR V30M patients compared to healthy controls; S100A8/9 levels in Schwann cells were dysregulated after incubation with human V30M TTR and by mutated bone marrow-derived macrophages in response to Toll-like receptor agonists. |
4.2. Mechanistic Insight from Animal Studies
| Reference | Animal Model | Methods and Techniques | Main Findings |
|---|---|---|---|
| Santos et al., 2010 [82] | V30M TTR/HSF1 mice vs. WT mice | SQ-IHC | Increase in pro-inflammatory cytokines TNF- α and IL1-β, and NF-kB activation occurring in dorsal roots ganglia. |
| Gonçalves et al., 2014 [83] | V30M TTR/HSF1 mice vs. WT mice | Flow cytometry and SQ-IHC | Downregulation of Cxcl-3, Cxcl-2, Cxcl-12, and TLR 1. Lower expressions of TNF-α and IL-1β. Upregulation of IL-10. No difference in the expression of IL-6. |
| Gonçalves et al., 2016 [89] | V30M TTR/HSF1 mice vs. WT mice | Microarray technology | TLR 1, Cxcl2, and Cxcl 3 were confirmed to be downregulated. |
| Moreira et al., 2021 [90] | V30M TTR/mice vs. WT mice | Real-time PCR | Decreased expressions of chemokines, such as Ccl20, Ccl8, Ccl5, Cxcl5, Ccl2, Cxcl2, and Cxcl3. Downregulation of IL-6. |
| Moreira et al., 2023 [91] | V30M TTR/mice vs. WT mice | Real-time PCR, cell culture | The expressions of several chemokines by bone marrow-derived macrophages generated from V30M TTR mice after stimulation with TLR4 and TLR2 agonists decreased; p38, which has a pivotal role for TLR4 and TLR2 signaling pathways, presented a reduced phosphorylation in V30M macrophages, compared to WT ones. |
| Gonçalves et al., 2015 [93] | V30M TTR/mice vs. WT mice | SQ-IHC; double immunofluorescence; immunogold labeling; real-time PCR; flow cytometry; Western blot; sciatic nerve morphometric analysis | Treatment with the IL-1 receptor antagonist Anakinra in FAP mice decreased inflammation markers and improved axonal non-myelinated fibers. |
| Buxbaum et al., 2012 [92] | Transgenic model expressing approximately 90 copies of the wild-type human TTR gene under the control of its own promoter | Transcriptomic analysis | Hepatic chaperone activity was deficient in mice with cardiac deposition; robust cardiac inflammatory response in 3-month-old mice who have no cardiac deposits, which changes in the hearts of 15–24-month-old mice with either fibrillar or non-fibrillar deposits. |
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luigetti, M.; Romano, A.; Di Paolantonio, A.; Bisogni, G.; Sabatelli, M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther. Clin. Risk Manag. 2020, 16, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2021, 268, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary transthyretin amyloidosis overview. Neurol. Sci. 2022, 43 (Suppl. 2), 595–604. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef]
- Suenaga, G.; Ikeda, T.; Masuda, T.; Motokawa, H.; Yamashita, T.; Takamatsu, K.; Misumi, Y.; Ueda, M.; Matsui, H.; Senju, S.; et al. Inflammatory State Exists in Familial Amyloid Polyneuropathy That May Be Triggered by Mutated Transthyretin. Sci. Rep. 2017, 7, 1579. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory Profiling of Patients with Familial Amyloid Polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romano, A.; Guglielmino, V.; Sciarrone, M.A.; Vitali, F.; Carbone, C.; Piro, G.; Sabino, A.; De Stefano, N.; Plantone, D.; et al. Serum Inflammatory Profile in Hereditary Transthyretin Amyloidosis: Mechanisms and Possible Therapeutic Implications. Brain Sci. 2022, 12, 1708. [Google Scholar] [CrossRef]
- Cras-Meneur, C.; Inoue, H.; Zhou, Y.; Ohsugi, M.; Bernal-Mizrachi, E.; Pape, D.; Clifton, S.W.; Permutt, M.A. An expression profile of human pancreatic islet mRNAs by Serial Analysis of Gene Expression (SAGE). Diabetologia 2004, 47, 284–299. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Reixach, N. Transthyretin: The Servant of Many Masters. Cell. Mol. Life Sci. 2009, 66, 3095–3101. [Google Scholar] [CrossRef]
- Ueda, M. Transthyretin: Its Function and Amyloid Formation. Neurochem. Int. 2022, 155, 3095–3101. [Google Scholar] [CrossRef]
- Palha, J.A. Transthyretin as a Thyroid Hormone Carrier: Function Revisited. Clin. Chem. Lab. Med. 2002, 40, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.F.; Geisow, M.J.; Swan, I.D.A.; Rerat, C.; Rerat, B. Structure of Human Plasma Prealbumin at 2.5 A Resolution. A Preliminary Report on the Polypeptide Chain Conformation, Quaternary Structure and Thyroxine Binding. J. Mol. Biol. 1974, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tozza, S.; Severi, D.; Spina, E.; Iovino, A.; Aruta, F.; Ruggiero, L.; Dubbioso, R.; Iodice, R.; Nolano, M.; Manganelli, F. The Neuropathy in Hereditary Transthyretin Amyloidosis: A Narrative Review. J. Peripher. Nerv. Syst. 2021, 26, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.B.; Coelho, T.; Waddington Cruz, M.; Brannagan, T.H.; Khella, S.; Karam, C.; Berk, J.L.; Polydefkis, M.J.; Kincaid, J.C.; Wiesman, J.F.; et al. Neuropathy Symptom and Change: Inotersen Treatment of Hereditary Transthyretin Amyloidosis. Muscle Nerve 2020, 62, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Coelho, T.; Rodrigues, A.; Felgueiras, H.; Oliveira, P.; Guimarães, A.; Melo-Pires, M.; Taipa, R. Clinicopathological Correlations of Sural Nerve Biopsies in TTR Val30Met Familial Amyloid Polyneuropathy. Brain Commun. 2019, 1, fcz032. [Google Scholar] [CrossRef] [PubMed]
- Skrahina, V.; Grittner, U.; Beetz, C.; Skripuletz, T.; Juenemann, M.; Krämer, H.H.; Hahn, K.; Rieth, A.; Schaechinger, V.; Patten, M.; et al. Hereditary Transthyretin-Related Amyloidosis Is Frequent in Polyneuropathy and Cardiomyopathy of No Obvious Aetiology. Ann. Med. 2021, 53, 1787–1796. [Google Scholar] [CrossRef]
- Sekijima, Y. Transthyretin (ATTR) Amyloidosis: Clinical Spectrum, Molecular Pathogenesis and Disease-Modifying Treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef]
- Carroll, A.; Dyck, P.J.; De Carvalho, M.; Kennerson, M.; Reilly, M.M.; Kiernan, M.C.; Vucic, S. Novel Approaches to Diagnosis and Management of Hereditary Transthyretin Amyloidosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 668–678. [Google Scholar] [CrossRef]
- Cortese, A.; Vegezzi, E.; Lozza, A.; Alfonsi, E.; Montini, A.; Moglia, A.; Merlini, G.; Obici, L. Diagnostic Challenges in Hereditary Transthyretin Amyloidosis with Polyneuropathy: Avoiding Misdiagnosis of a Treatable Hereditary Neuropathy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 457–458. [Google Scholar] [CrossRef]
- Mathis, S.; Magy, L.; Diallo, L.; Boukhris, S.; Vallat, J.M. Amyloid Neuropathy Mimicking Chronic Inflammatory Demyelinating Polyneuropathy. Muscle Nerve 2012, 45, 26–31. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Said, G. Familial Amyloid Polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Vera-Llonch, M.; Reddy, S.R.; Chang, E.; Tarbox, M.H.; Pollock, M. The Patient Journey toward a Diagnosis of Hereditary Transthyretin (ATTRv) Amyloidosis. Orphanet. J. Rare Dis. 2021, 16, 25. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol Ther. 2020, 9, 317–333. [Google Scholar] [CrossRef]
- Stewart, M.; Shaffer, S.; Murphy, B.; Loftus, J.; Alvir, J.; Cicchetti, M.; Lenderking, W.R. Characterizing the High Disease Burden of Transthyretin Amyloidosis for Patients and Caregivers. Neurol. Ther. 2018, 7, 349–364. [Google Scholar] [CrossRef]
- Yarlas, A.; Gertz, M.A.; Dasgupta, N.R.; Obici, L.; Pollock, M.; Ackermann, E.J.; Lovley, A.; Kessler, A.S.; Patel, P.A.; White, M.K.; et al. Burden of Hereditary Transthyretin Amyloidosis on Quality of Life. Muscle Nerve 2019, 60, 169–175. [Google Scholar] [CrossRef]
- Tozza, S.; Luigetti, M.; Antonini, G.; Mazzeo, A.; Severi, D.; Di Paolantonio, A.; Leonardi, L.; Russo, M.; Romano, A.; Forcina, F.; et al. Neuropathic Pain Experience in Symptomatic and Presymptomatic Subjects Carrying a Transthyretin Gene Mutation. Front. Neurol. 2023, 14, 1109782. [Google Scholar] [CrossRef]
- Coelho, T.; Waddington Cruz, M.; Chao, C.C.; Parman, Y.; Wixner, J.; Weiler, M.; Barroso, F.A.; Dasgupta, N.R.; Jung, S.W.; Schneider, E.; et al. Characteristics of Patients with Hereditary Transthyretin Amyloidosis-Polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an Open-Label Phase 3 Study of Eplontersen. Neurol. Ther. 2023, 12, 267–287. [Google Scholar] [CrossRef]
- Benbrahim, M.; Norman, K.; Sanchorawala, V.; Siddiqi, O.K.; Hughes, D. A Review of Novel Agents and Clinical Considerations in Patients with ATTR Cardiac Amyloidosis. J. Cardiovasc. Pharmacol. 2021, 77, 544–548. [Google Scholar] [CrossRef]
- Ferraro, P.M.; D’Ambrosio, V.; Di Paolantonio, A.; Guglielmino, V.; Calabresi, P.; Sabatelli, M.; Luigetti, M. Renal Involvement in Hereditary Transthyretin Amyloidosis: An Italian Single-Centre Experience. Brain Sci. 2021, 11, 980. [Google Scholar] [CrossRef]
- Ando, Y.; Adams, D.; Benson, M.D.; Berk, J.L.; Planté-Bordeneuve, V.; Coelho, T.; Conceição, I.; Ericzon, B.G.; Obici, L.; Rapezzi, C.; et al. Guidelines and New Directions in the Therapy and Monitoring of ATTRv Amyloidosis. Amyloid 2022, 29, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Kyriakides, T. Drug and Gene Therapy for Treating Variant Transthyretin Amyloidosis (ATTRv) Neuropathy. Curr. Neuropharmacol. 2022, 21, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, G.; Steen, L.; Suhr, O.; Ericzon, B.G.; Groth, C.G.; Andersen, O.; Wallin, B.G.; Seymour, A.; Richardson, S.; Hawkins, P.N.; et al. Clinical Improvement and Amyloid Regression after Liver Transplantation in Hereditary Transthyretin Amyloidosis. Lancet 1993, 341, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Wixner, J.; Obayashi, K.; Ando, Y.; Ericzon, B.G.; Friman, S.; Uchino, M.; Suhr, O.B. Liver Transplantation for Familial Amyloidotic Polyneuropathy: Impact on Swedish Patients’ Survival. Liver Transplant. 2009, 15, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, H.E.; Larsson, M.; Ericzon, B.G. Long-Term Data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid 2011, 18 (Suppl. 1), 193–195. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S.I. Safety and Efficacy of Long-Term Diflunisal Administration in Hereditary Transthyretin (ATTR) Amyloidosis. Amyloid 2015, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Planté-Bordeneuve, V.; Judge, D.P.; Schmidt, H.; Obici, L.; Perlini, S.; Packman, J.; Tripp, T.; Grogan, D.R. Effects of Tafamidis on Transthyretin Stabilization and Clinical Outcomes in Patients with Non-Val30Met Transthyretin Amyloidosis. J. Cardiovasc. Transl. Res. 2013, 6, 1011–1120. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Benson, M.D.; Dasgupta, N.R.; Rissing, S.M.; Smith, J.; Feigenbaum, H. Safety and Efficacy of a TTR Specific Antisense Oligonucleotide in Patients with Transthyretin Amyloid Cardiomyopathy. Amyloid 2017, 24, 219–225. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2023, 30, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Sakurai, T.; Shimohata, T.; Katsuno, M.; Sobue, G. The Morphology of Amyloid Fibrils and Their Impact on Tissue Damage in Hereditary Transthyretin Amyloidosis: An Ultrastructural Study. J. Neurol. Sci. 2018, 394, 99–106. [Google Scholar] [CrossRef]
- Koike, H.; Ikeda, S.; Takahashi, M.; Kawagashira, Y.; Iijima, M.; Misumi, Y.; Ando, Y.; Ikeda, S.I.; Katsuno, M.; Sobue, G. Schwann Cell and Endothelial Cell Damage in Transthyretin Familial Amyloid Polyneuropathy. Neurology 2016, 87, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, A.; Andrade, C. Familial Amyloid Polyneuropathy: An Electron Microscope Study of the Peripheral Nerve in Five Cases. I. Interstitial Changes. Brain 1971, 94, 199–206. [Google Scholar] [CrossRef]
- Ando, Y.; Nyhlin, N.; Suhr, O.; Holmgren, G.; Uchida, K.; El Sahly, M.; Yamashita, T.; Terasaki, H.; Nakamura, M.; Uchino, M.; et al. Oxidative Stress Is Found in Amyloid Deposits in Systemic Amyloidosis. Biochem. Biophys. Res. Commun. 1997, 232, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Misu, K.; Sugiura, M.; Iijima, M.; Mori, K.; Yamamoto, M.; Hattori, N.; Mukai, E.; Ando, Y.; Ikeda, S.; et al. Pathology of Early- vs Late-Onset TTR Met30 Familial Amyloid Polyneuropathy. Neurology 2004, 63, 129–138. [Google Scholar] [CrossRef]
- Ihse, E.; Ybo, A.; Suhr, O.B.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid Fibril Composition Is Related to the Phenotype of Hereditary Transthyretin V30M Amyloidosis. J. Pathol. 2008, 216, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J.; Magalhaes, J.; Ferreira, N.; Almeida, M.R. Transthyretin deposition in familial amyloidotic polyneuropathy. Curr. Med. Chem. 2012, 19, 2304–2311. [Google Scholar] [CrossRef]
- Sousa, M.M.; Saraiva, M.J. Neurodegeneration in Familial Amyloid Polyneuropathy: From Pathology to Molecular Signaling. Prog. Neurobiol. 2003, 71, 385–400. [Google Scholar] [CrossRef]
- Sousa, M.M.; Ferraõ, J.; Fernandes, R.; Guimarães, A.; Geraldes, J.B.; Perdigoto, R.; Tomé, L.; Mota, O.; Negrão, L.; Furtado, A.L.; et al. Deposition and Passage of Transthyretin through Blood-Nerve Barrier in Recipients of Familial Amyloid Polyneuropathy Livers. Lab. Investig. 2004, 84, 865–873. [Google Scholar] [CrossRef]
- Macedo, B.; Batista, A.R.; do Amaral, J.B.; Saraiva, M.J. Biomarkers in the assessment of therapies for familial amyloidotic polyneuropathy. Mol. Med. 2007, 13, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Yan, S.D.; Fernandas, R.; Guimarães, A.; Stern, D.; Saraiva, M.J. Familial Amyloid Polyneuropathy: Receptor for Advanced Glycation End Products-Dependent Triggering of Neuronal Inflammatory and Apoptotic Pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Anan, I.; Forsgren, S.; Nagai, R.; Rosenberg, P.; Horiuchi, S.; Ando, Y.; Suhr, O.B. Advanced Glycation End Products (AGE) and the Receptor for AGE Are Present in Gastrointestinal Tract of Familial Amyloidotic Polyneuropathy Patients but Do Not Induce NF-ΚB Activation. Acta Neuropathol. 2002, 104, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Said, G.; Ropert, A.; Faux, N. Length-dependent degeneration of fibers in Portuguese amyloid polyneuropathy: A clinicopathologic study. Neurology 1984, 34, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.E.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.; Ray, R.; Banach, M.; Rai, V. Receptor for Advanced Glycation End-Products in Neurodegenerative Diseases. Rev. Neurosci. 2015, 26, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Yan, S.D.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s disease: A progression factor for amyloid-beta-induced cellular perturbation? J. Alzheimers Dis. 2009, 16, 833–843. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic β cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Sbai, O.; Devi, T.S.; Melone, M.A.B.; Feron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP Axis Is Required for S100B-Promoted Schwann Cell Migration, Fibronectin Expression and Cytokine Secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef]
- Juranek, J.K.; Kothary, P.; Mehra, A.; Hays, A.; Brannagan, T.H.; Schmidt, A.M. Increased Expression of the Receptor for Advanced Glycation End-Products in Human Peripheral Neuropathies. Brain Behav. 2013, 3, 701–709. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, Y.-A.; Hong, Y.-H.; Moon, M.-K.; Koo, B.-K.; Kim, T.W. Up-Regulation of the Receptor for Advanced Glycation End Products in the Skin Biopsy Specimens of Patients with Severe Diabetic Neuropathy. J. Clin. Neurol. 2014, 10, 334. [Google Scholar] [CrossRef]
- Bekircan-Kurt, C.E.; Üçeyler, N.; Sommer, C. Cutaneous Activation of Rage in Nonsystemic Vasculitic and Diabetic Neuropathy. Muscle Nerve 2014, 50, 377–383. [Google Scholar] [CrossRef]
- Yan, S.D.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu, J.; Soto, C.; Schmidt, A.M.; Stern, D.; et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat. Med. 2000, 6, 643–651. [Google Scholar] [CrossRef]
- Moreira, J.; Martins, S.; Saraiva, M.; Saraiva, M.J. Decreased expression of S100A8/A9 in V30M related ATTRv amyloidosis. Amyloid 2023, 30, 1–8. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.C. NF-ΚB in Inflammation and Renal Diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The I Kappa B Kinase (IKK) and NF-Kappa B: Key Elements of Proinflammatory Signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-ΚB: Roles and Regulation in Different CD4+ T-Cell Subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef]
- Sun, S.C. The Noncanonical NF-ΚB Pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Sun, S.C.; Liu, Z.G. A Special Issue on NF-ΚB Signaling and Function. Cell Res. 2011, 21, 1–2. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A Critical Review of Its Biology and the Mechanisms Involved in Its Release as a Potent Extracellular Cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in Aging and Chronic Disease: A Magnificent Pathway. J. Gerontol. Ser. A 2006, 61, 575–584. [Google Scholar] [CrossRef]
- Kurian, S.M.; Novais, M.; Whisenant, T.; Gelbart, T.; Buxbaum, J.N.; Kelly, J.W.; Coelho, T.; Salomon, D.R. Peripheral Blood Cell Gene Expression Diagnostic for Identifying Symptomatic Transthyretin Amyloidosis Patients: Male and Female Specific Signatures. Theranostics 2016, 6, 1792–1809. [Google Scholar] [CrossRef]
- Fontana, M.; Gilbertson, J.; Verona, G.; Riefolo, M.; Slamova, I.; Leone, O.; Rowczenio, D.; Botcher, N.; Ioannou, A.; Patel, R.K.; et al. Antibody-Associated Reversal of ATTR Amyloidosis-Related Cardiomyopathy. N. Engl. J. Med. 2023, 388, 2199–2201. [Google Scholar] [CrossRef]
- Michalon, A.; Hagenbuch, A.; Huy, C.; Varela, E.; Combaluzier, B.; Damy, T.; Suhr, O.B.; Saraiva, M.J.; Hock, C.; Nitsch, R.M.; et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021, 12, 3142. [Google Scholar] [CrossRef]
- Phay, M.; Blinder, V.; Macy, S.; Greene, M.J.; Wooliver, D.C.; Liu, W.; Planas, A.; Walsh, D.M.; Connors, L.H.; Primmer, S.R.; et al. Transthyretin aggregate-specific antibodies recognize cryptic epitopes on patient-derived amyloid fibrils. Rejuvenation Res. 2014, 17, 97–104. [Google Scholar] [CrossRef]
- Faravelli, G.; Mondani, V.; Mangione, P.P.; Raimondi, S.; Marchese, L.; Lavatelli, F.; Stoppini, M.; Corazza, A.; Canetti, D.; Verona, G.; et al. Amyloid Formation by Globular Proteins: The Need to Narrow the Gap Between in Vitro and in Vivo Mechanisms. Front. Mol. Biosci. 2022, 9, 830006. [Google Scholar] [CrossRef]
- Yi, S.; Takahashi, K.; Naito, M.; Tashiro, F.; Wakasugi, S.; Maeda, S.; Shimada, K.; Yamamura, K.; Araki, S. Systemic Amyloidosis in Transgenic Mice Carrying the Human Mutant Transthyretin (Met30) Gene. Pathologic Similarity to Human Familial Amyloidotic Polyneuropathy, Type I. Am. J. Pathol. 1991, 138, 403–412. [Google Scholar]
- Santos, S.D.; Fernandes, R.; Saraiva, M.J. The Heat Shock Response Modulates Transthyretin Deposition in the Peripheral and Autonomic Nervous Systems. Neurobiol. Aging 2010, 31, 280–289. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Teixeira-Coelho, M.; Saraiva, M.J. The Inflammatory Response to Sciatic Nerve Injury in a Familial Amyloidotic Polyneuropathy Mouse Model. Exp. Neurol. 2014, 257, 76–87. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 Contributes to Inflammatory and Neuropathic Pain through the Aggravation of Pronociceptive Inflammatory Responses in Mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef]
- Smith, D.F.; Galkina, E.; Ley, K.; Huo, Y. GRO Family Chemokines Are Specialized for Monocyte Arrest from Flow. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1976–H1984. [Google Scholar] [CrossRef]
- Heskamp, A.; Leibinger, M.; Andreadaki, A.; Gobrecht, P.; Diekmann, H.; Fischer, D. CXCL12/SDF-1 Facilitates Optic Nerve Regeneration. Neurobiol. Dis. 2013, 55, 76–86. [Google Scholar] [CrossRef]
- Nyhlin, N.; Ando, Y.; Nagai, R.; Suhr, O.; El Sahly, M.; Terazaki, H.; Yamashita, T.; Ando, M.; Horiuchi, S. Advanced Glycation End Product in Familial Amyloidotic Polyneuropathy (FAP). J. Intern. Med. 2000, 247, 485–492. [Google Scholar] [CrossRef]
- Misu, K.I.; Hattori, N.; Nagamatsu, M.; Ikeda, S.I.; Ando, Y.; Nakazato, M.; Takei, Y.I.; Hanyu, N.; Usui, Y.; Tanaka, F.; et al. Late-Onset Familial Amyloid Polyneuropathy Type I (Transthyretin Met30-Associated Familial Amyloid Polyneuropathy) Unrelated to Endemic Focus in Japan. Clinicopathological and Genetic Features. Brain 1999, 122, 1951–1962. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Martins, D.; Saraiva, M.J. Overexpression of Protocadherin-10 in Transthyretin-Related Familial Amyloidotic Polyneuropathy. Am. J. Pathol. 2016, 186, 1913–1924. [Google Scholar] [CrossRef]
- Moreira, J.; Costelha, S.; Saraiva, M.; Saraiva, M.J. The Expression of Chemokines Is Downregulated in a Pre-Clinical Model of TTR V30M Amyloidosis. Front. Immunol. 2021, 12, 1823. [Google Scholar] [CrossRef]
- Moreira, J.; Martins, H.; Saraiva, M.; Saraiva, M.J. TLR2 and 4 signaling pathways are altered in macrophages from V30M TTR mice with down-regulated expression of chemokines. Clin. Sci. 2023, 137, 355–366. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Tagoe, C.; Gallo, G.; Walker, J.R.; Kurian, S.; Salomon, D.R. Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012, 26, 2283–2293. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Teixeira-Coelho, M.; Saraiva, M.J. Protective role of anakinra against transthyretin-mediated axonal loss and cell death in a mouse model of familial amyloidotic polyneuropathy. J. Neuropathol. Exp. Neurol. 2015, 74, 203–217. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plantone, D.; Primiano, G.; Righi, D.; Romano, A.; Luigetti, M.; De Stefano, N. Current Evidence Supporting the Role of Immune Response in ATTRv Amyloidosis. Cells 2023, 12, 2383. https://doi.org/10.3390/cells12192383
Plantone D, Primiano G, Righi D, Romano A, Luigetti M, De Stefano N. Current Evidence Supporting the Role of Immune Response in ATTRv Amyloidosis. Cells. 2023; 12(19):2383. https://doi.org/10.3390/cells12192383
Chicago/Turabian StylePlantone, Domenico, Guido Primiano, Delia Righi, Angela Romano, Marco Luigetti, and Nicola De Stefano. 2023. "Current Evidence Supporting the Role of Immune Response in ATTRv Amyloidosis" Cells 12, no. 19: 2383. https://doi.org/10.3390/cells12192383
APA StylePlantone, D., Primiano, G., Righi, D., Romano, A., Luigetti, M., & De Stefano, N. (2023). Current Evidence Supporting the Role of Immune Response in ATTRv Amyloidosis. Cells, 12(19), 2383. https://doi.org/10.3390/cells12192383