

Differential Susceptibility of Ex Vivo Primary Glioblastoma Tumors to Oncolytic Effect of Modified Zika Virus
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Lines
2.3. Zika Virus
2.4. Zika Viral Infection
2.5. Quantification of ZIKV with Plaque Assay
2.6. Mouse Experiment
2.7. Mouse GBM Xenograft Study
2.8. Immunohistochemistry
2.9. Western Blot Analysis
2.10. Data Analysis
2.11. Data Availability
3. Results
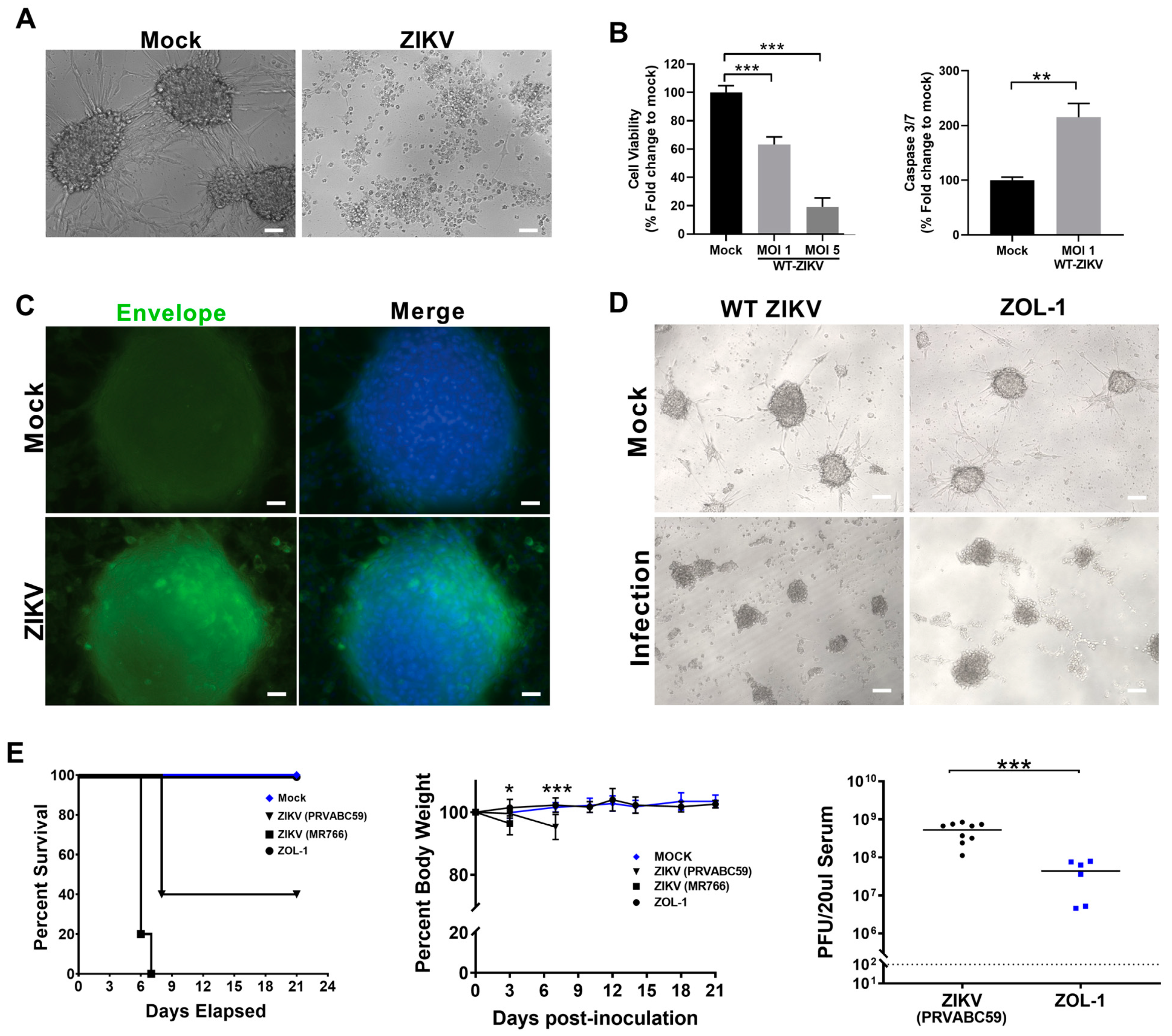
3.1. Attenuated ZIKV Strain ZOL-1 Targets GSCs and Is Safe in Mice
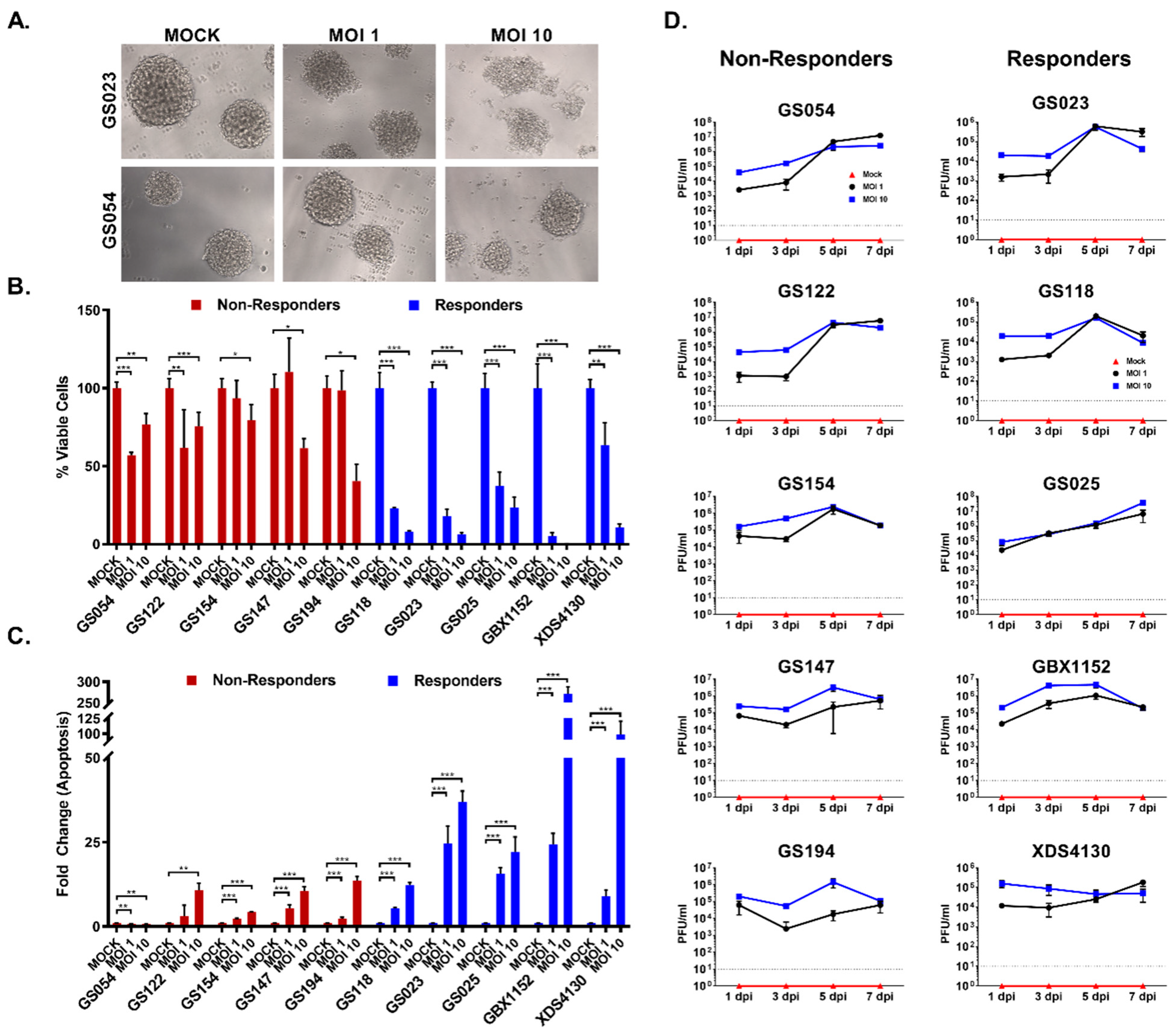
3.2. GBM Cell Lines Respond Differently to ZOL-1 Infection
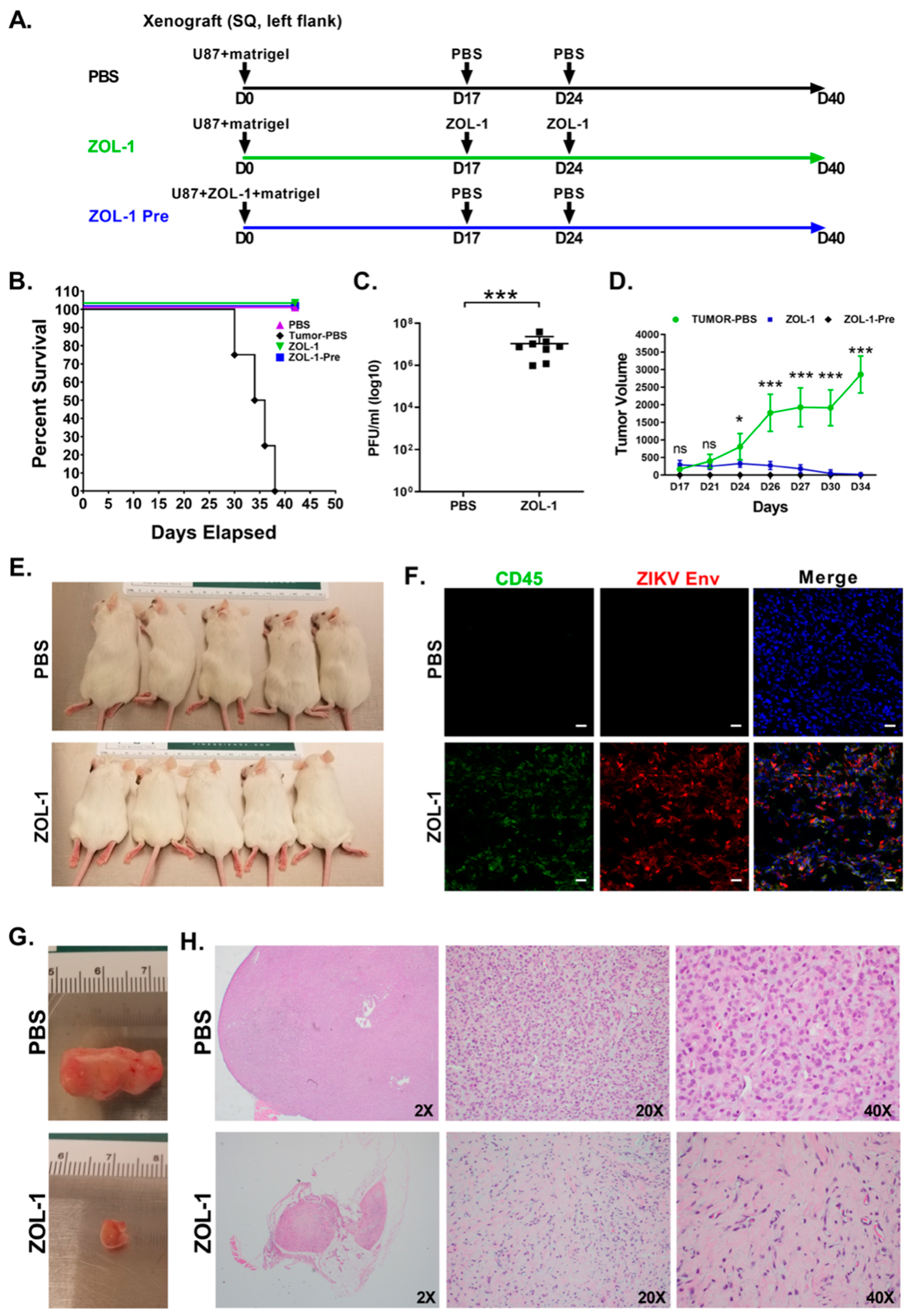
3.3. ZOL-1 Displays Clear Antitumor Effects on Xenograft NSG Mice
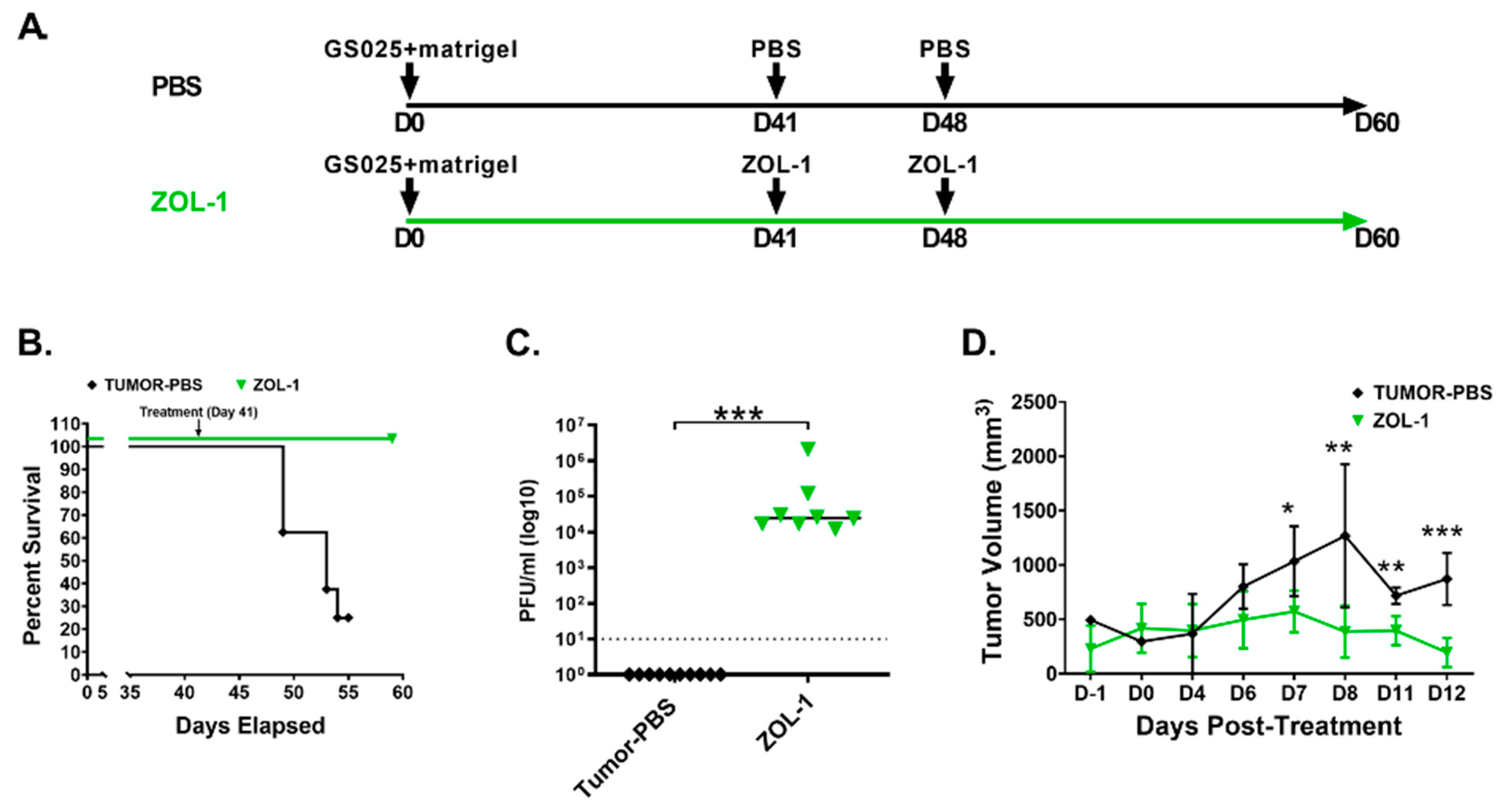
3.4. ZOL-1 Displays Effective Antitumor Action against Human GBM-Derived Cell Lines
3.5. Combination Therapies Enhance ZOL-1 Effects on Non-Responder GBM Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Omuro, A. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Zhou, Q.; Xue, C.; Ke, X.; Zhou, J. Treatment Response and Prognosis Evaluation in High-Grade Glioma: An Imaging Review Based on MRI. Magn. Reson. Imaging 2022, 56, 325–340. [Google Scholar] [CrossRef]
- Ma, Q.; Long, W.; Xing, C.; Chu, J.; Luo, M.; Wang, H.Y.; Liu, Q.; Wang, R.-F. Cancer Stem Cells and Immunosuppressive Microenvironment in Glioma. Front. Immunol. 2018, 9, 2924. [Google Scholar] [CrossRef]
- Sugiarto, S.; Persson, A.I.; Munoz, E.G.; Waldhuber, M.; Lamagna, C.; Andor, N.; Hanecker, P.; Ayers-Ringler, J.; Phillips, J.; Siu, J.; et al. Asymmetry-Defective Oligodendrocyte Progenitors Are Glioma Precursors. Cancer Cell 2011, 20, 328–340. [Google Scholar] [CrossRef]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Davis, M. Glioblastoma: Overview of Disease and Treatment. CJON 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Frederico, S.C.; Hancock, J.C.; Brettschneider, E.E.S.; Ratnam, N.M.; Gilbert, M.R.; Terabe, M. Making a Cold Tumor Hot: The Role of Vaccines in the Treatment of Glioblastoma. Front. Oncol. 2021, 11, 672508. [Google Scholar] [CrossRef]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti–PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. NEUONC 2016, 19, 493–502. [Google Scholar] [CrossRef]
- Bach, P.; Abel, T.; Hoffmann, C.; Gal, Z.; Braun, G.; Voelker, I.; Ball, C.R.; Johnston, I.C.D.; Lauer, U.M.; Herold-Mende, C.; et al. Specific Elimination of CD133+ Tumor Cells with Targeted Oncolytic Measles Virus. Cancer Res. 2013, 73, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Opyrchal, M.; Dispenzieri, A.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; Galanis, E. Clinical Trials with Oncolytic Measles Virus: Current Status and Future Prospects. CCDT 2018, 18, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sarkaria, J.N.; Petell, C.A.; Paraskevakou, G.; Zollman, P.J.; Schroeder, M.; Carlson, B.; Decker, P.A.; Wu, W.; James, C.D.; et al. Combination of Measles Virus Virotherapy and Radiation Therapy Has Synergistic Activity in the Treatment of Glioblastoma Multiforme. Clin. Cancer Res. 2007, 13, 7155–7165. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Dobrikova, E.Y.; Broadt, T.; Poiley-Nelson, J.; Yang, X.; Soman, G.; Giardina, S.; Harris, R.; Gromeier, M. Recombinant Oncolytic Poliovirus Eliminates Glioma In Vivo Without Genetic Adaptation to a Pathogenic Phenotype. Mol. Ther. 2008, 16, 1865–1872. [Google Scholar] [CrossRef]
- Huang, J.; LaRocca, C.; Yamamoto, M. Showing the Way: Oncolytic Adenoviruses as Chaperones of Immunostimulatory Adjuncts. Biomedicines 2016, 4, 23. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model: KURYK. J. Med. Virol. 2018, 90, 1669–1673. [Google Scholar] [CrossRef]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2019, 37, 45–54. [Google Scholar] [CrossRef]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi–mutated herpes simplex virus–1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Burton, C.; Das, A.; McDonald, D.; Vandergrift, W.A., III; Patel, S.J.; Cachia, D.; Bartee, E. Oncolytic myxoma virus synergizes with standard of care for treatment of glioblastoma multiforme. OV 2018, 7, 107–116. [Google Scholar] [CrossRef]
- Zemp, F.J.; Lun, X.; McKenzie, B.A.; Zhou, H.; Maxwell, L.; Sun, B.; Kelly, J.J.P.; Stechishin, O.; Luchman, A.; Weiss, S.; et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro-Oncol. 2013, 15, 904–920. [Google Scholar] [CrossRef]
- Wollmann, G.; Rogulin, V.; Simon, I.; Rose, J.K.; van den Pol, A.N. Some Attenuated Variants of Vesicular Stomatitis Virus Show Enhanced Oncolytic Activity against Human Glioblastoma Cells relative to Normal Brain Cells. J. Virol. 2010, 84, 1563–1573. [Google Scholar] [CrossRef]
- Markert, J.M. Safety and Efficacy Study of REOLYSIN® in the Treatment of Recurrent Malignant Gliomas; 2014. Available online: https://clinicaltrials.gov/study/NCT00528684 (accessed on 9 September 2023).
- Unterberg, A. Parvovirus H-1 (ParvOryx) in Patients with Progressive Primary or Recurrent Glioblastoma Multiforme (ParvOryx01); 2015. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT01301430 (accessed on 9 September 2023).
- Landi, D.; Thompson, E. Phase 1b Study PVSRIPO for Recurrent Malignant Glioma in Children; 2017. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03043391 (accessed on 9 September 2023).
- Home—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/home (accessed on 9 September 2023).
- Greig, S.L. Talimogene Laherparepvec: First Global Approval. Drugs 2016, 76, 147–154. [Google Scholar] [CrossRef]
- Jaime-Ramirez, A.C.; Yu, J.-G.; Caserta, E.; Yoo, J.Y.; Zhang, J.; Lee, T.J.; Hofmeister, C.; Lee, J.H.; Kumar, B.; Pan, Q.; et al. Reolysin and Histone Deacetylase Inhibition in the Treatment of Head and Neck Squamous Cell Carcinoma. Mol. Ther.-Oncolytics 2017, 5, 87–96. [Google Scholar] [CrossRef]
- Ribeiro, G.S.; Kitron, U. Zika virus pandemic: A human and public health crisis. Rev. Soc. Bras. Med. Trop. 2016, 49, 1–3. [Google Scholar] [CrossRef]
- Imperato, P.J. The Convergence of a Virus, Mosquitoes, and Human Travel in Globalizing the Zika Epidemic. J. Community Health 2016, 41, 674–679. [Google Scholar] [CrossRef]
- Kakooza-Mwesige, A.; Tshala-Katumbay, D.; Juliano, S.L. Viral infections of the central nervous system in Africa. Brain Res. Bull. 2019, 145, 2–17. [Google Scholar] [CrossRef]
- Carod-Artal, F.J. Neurological complications of Zika virus infection. Expert. Rev. Anti-Infect. Ther. 2018, 16, 399–410. [Google Scholar] [CrossRef]
- Russo, F.B.; Beltrão-Braga, P.C.B. The impact of Zika virus in the brain. Biochem. Biophys. Res. Commun. 2017, 492, 603–607. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef]
- Bayless, N.L.; Greenberg, R.S.; Swigut, T.; Wysocka, J.; Blish, C.A. Zika Virus Infection Induces Cranial Neural Crest Cells to Produce Cytokines at Levels Detrimental for Neurogenesis. Cell Host Microbe 2016, 20, 423–428. [Google Scholar] [CrossRef]
- Ming, G.; Tang, H.; Song, H. Advances in Zika Virus Research: Stem Cell Models, Challenges, and Opportunities. Cell Stem Cell 2016, 19, 690–702. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.-H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant Astrocytomas Originate from Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of Human Glioblastoma with a Live Attenuated Zika Virus Vaccine Candidate. mBio 2018, 9, e01683-18. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, C.; Chen, Q.; Shang, J.; Liu, Z.; Guo, Y.; Li, C.; Wang, H.; Ye, Q.; Li, X.; et al. Oncolytic Zika virus promotes intratumoral T cell infiltration and improves immunotherapy efficacy in glioblastoma. Mol. Ther.-Oncolytics 2022, 24, 522–534. [Google Scholar] [CrossRef]
- Trus, I.; Berube, N.; Jiang, P.; Rak, J.; Gerdts, V.; Karniychuk, U. Zika Virus with Increased CpG Dinucleotide Frequencies Shows Oncolytic Activity in Glioblastoma Stem Cells. Viruses 2020, 12, 579. [Google Scholar] [CrossRef]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin αvβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef]
- Lubin, J.A.; Zhang, R.R.; Kuo, J.S. Zika Virus has Oncolytic Activity Against Glioblastoma Stem Cells. Neurosurgery 2018, 82, E113–E114. [Google Scholar] [CrossRef] [PubMed]
- Kaid, C.; Goulart, E.; Caires-Júnior, L.C.; Araujo, B.H.S.; Soares-Schanoski, A.; Bueno, H.M.S.; Telles-Silva, K.A.; Astray, R.M.; Assoni, A.F.; Júnior, A.F.R.; et al. Zika Virus Selectively Kills Aggressive Human Embryonal CNS Tumor Cells In Vitro and In Vivo. Cancer Res. 2018, 78, 3363–3374. [Google Scholar] [CrossRef]
- Arumugaswami, V.; Breunig, J. Methods of Inducing an Oncolytic Effect on Tumor Cells Using Zika Virus. U.S. Patent Application No. 16/322,396, 27 June 2019. [Google Scholar]
- Aspirin, A.P.; De Los Reyes V, A.A.; Kim, Y. Polytherapeutic strategies with oncolytic virus–bortezomib and adjuvant NK cells in cancer treatment. J. R. Soc. Interface. 2021, 18, 20200669. [Google Scholar] [CrossRef]
- Alvarez-Breckenridge, C.; Kaur, B.; Chiocca, E.A. Pharmacologic and Chemical Adjuvants in Tumor Virotherapy. Chem. Rev. 2009, 109, 3125–3140. [Google Scholar] [CrossRef]
- Terrível, M.; Gromicho, C.; Matos, A.M. Oncolytic viruses: What to expect from their use in cancer treatment. Microbiol. Immunol. 2020, 64, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Mai, W.X.; Gosa, L.; Daniels, V.W.; Ta, L.; Tsang, J.E.; Higgins, B.; Gilmore, W.B.; Bayley, N.A.; Harati, M.D.; Lee, J.T.; et al. Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat. Med. 2017, 23, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.E.; Garcia, G.; Contreras, D.; Gong, D.; Sun, R.; Arumugaswami, V. Zika Virus Mucosal Infection Provides Protective Immunity. J. Virol. 2020, 94, e00067-20. [Google Scholar] [CrossRef]
- Lemke, L.E.; Paine-Murrieta, G.D.; Taylor, C.W.; Powis, G. Wortmannin inhibits the growth of mammary tumors despite the existence of a novel wortmannin-insensitive phosphatidylinositol-3-kinase. Cancer Chemother. Pharmacol. 1999, 44, 491–497. [Google Scholar] [CrossRef]
- Breznik, B.; Ko, M.-W.; Tse, C.; Chen, P.-C.; Senjor, E.; Majc, B.; Habič, A.; Angelillis, N.; Novak, M.; Župunski, V.; et al. Infiltrating natural killer cells bind, lyse and increase chemotherapy efficacy in glioblastoma stem-like tumorospheres. Commun. Biol. 2022, 5, 436. [Google Scholar] [CrossRef]
- Gong, D.; Zhang, T.-H.; Zhao, D.; Du, Y.; Chapa, T.J.; Shi, Y.; Wang, L.; Contreras, D.; Zeng, G.; Shi, P.-Y.; et al. High-Throughput Fitness Profiling of Zika Virus E Protein Reveals Different Roles for Glycosylation during Infection of Mammalian and Mosquito Cells. iScience 2018, 1, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J.; et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Pourzia, A.L.; Nourian, A.A.; Williams, K.J.; Nathanson, D.; Babic, I.; Villa, G.R.; Tanaka, K.; Nael, A.; Yang, H.; et al. De-repression of PDGFRβ transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 2013, 3, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.R.; Nathanson, D.; Ponte de Albuquerque, C.; Kuga, D.; Iwanami, A.; Dang, J.; Yang, H.; Tanaka, K.; Oba-Shinjo, S.M.; Uno, M.; et al. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc. Natl. Acad. Sci. USA 2012, 109, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef]
- Yoshida, K.; Wilkins, J.; Winkler, J.; Wade, J.R.; Kotani, N.; Wang, N.; Sane, R.; Chanu, P. Population Pharmacokinetics of Ipatasertib and Its Metabolite in Cancer Patients. J. Clin. Pharmacol. 2021, 61, 1579–1591. [Google Scholar] [CrossRef]
- Sun, D.; Sawada, A.; Nakashima, M.; Kobayashi, T.; Ogawa, O.; Matsui, Y. MK2206 potentiates cisplatin-induced cytotoxicity and apoptosis through an interaction of inactivated Akt signaling pathway. Urol. Oncol. 2015, 33, 111.e17–111.e26. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296 Pt 2, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Nakanishi, S.; Kimura, K.; Hanai, N.; Saitoh, Y.; Fukui, Y.; Nonomura, Y.; Matsuda, Y. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in RBL-2H3 cells. J. Biol. Chem. 1993, 268, 25846–25856. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar]
- Moore, A.E. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer 1949, 2, 525–534. [Google Scholar] [CrossRef]
- Moore, A.E. Effect of inoculation of the viruses of influenza A and herpes simplex on the growth of transplantable tumors in mice. Cancer 1949, 2, 516–524. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef]
- Rajaraman, S.; Canjuga, D.; Ghosh, M.; Codrea, M.C.; Sieger, R.; Wedekink, F.; Tatagiba, M.; Koch, M.; Lauer, U.M.; Nahnsen, S.; et al. Measles Virus-Based Treatments Trigger a Pro-inflammatory Cascade and a Distinctive Immunopeptidome in Glioblastoma. Mol. Ther.-Oncolytics 2019, 12, 147–161. [Google Scholar] [CrossRef]
- Martinez-Velez, N.; Marigil, M.; García-Moure, M.; Gonzalez-Huarriz, M.; Aristu, J.J.; Ramos-García, L.-I.; Tejada, S.; Díez-Valle, R.; Patiño-García, A.; Becher, O.J.; et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol. Commun. 2019, 7, 64. [Google Scholar] [CrossRef]
- González-Morales, A.; Zabaleta, A.; García-Moure, M.; Alonso, M.M.; Fernández-Irigoyen, J.; Santamaría, E. Oncolytic adenovirus Delta-24-RGD induces a widespread glioma proteotype remodeling during autophagy. J. Proteom. 2019, 194, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, Q.; Chen, Y.; Qin, C.-F. Oncolytic Zika Virus: New Option for Glioblastoma Treatment. DNA Cell Biol. 2022, 42, 267–273. [Google Scholar] [CrossRef]
- Francipane, M.G.; Douradinha, B.; Chinnici, C.M.; Russelli, G.; Conaldi, P.G.; Iannolo, G. Zika Virus: A New Therapeutic Candidate for Glioblastoma Treatment. Int. J. Mol. Sci. 2021, 22, 10996. [Google Scholar] [CrossRef] [PubMed]
- Jash, A.; Govero, J.; Nair, S.; Diamond, M.S.; Chheda, M.G. Abstract A13: Leveraging Zika virus and the immune system to treat glioblastoma. Cancer Immunol. Res. 2020, 8, A13. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Rahman, M.; Sawyer, W.G.; Lindhorst, S.; Deleyrolle, L.P.; Harrison, J.K.; Karachi, A.; Dastmalchi, F.; Flores-Toro, J.; Mitchell, D.A.; Lim, M.; et al. Adult immuno-oncology: Using past failures to inform the future. Neuro Oncol. 2020, 22, 1249–1261. [Google Scholar] [CrossRef]
- O’Brien, C.; Wallin, J.J.; Sampath, D.; GuhaThakurta, D.; Savage, H.; Punnoose, E.A.; Guan, J.; Berry, L.; Prior, W.W.; Amler, L.C.; et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3’ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin. Cancer Res. 2010, 16, 3670–3683. [Google Scholar] [CrossRef]
- De Wolf, E.; De Wolf, C.; Richardson, A. ABT-737 and pictilisib synergistically enhance pitavastatin-induced apoptosis in ovarian cancer cells. Oncol. Lett. 2018, 15, 1979–1984. [Google Scholar] [CrossRef]
- Schöffski, P.; Cresta, S.; Mayer, I.A.; Wildiers, H.; Damian, S.; Gendreau, S.; Rooney, I.; Morrissey, K.M.; Spoerke, J.M.; Ng, V.W.; et al. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res. 2018, 20, 109. [Google Scholar] [CrossRef]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Mayer, I.A.; Ganju, V.; Dickler, M.; Johnston, S.; Morales, S.; Yardley, D.A.; Melichar, B.; Forero-Torres, A.; Lee, S.C.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Dent, R.A.; O’Shaughnessy, J.; Kim, S.-B.; Isakoff, S.J.; Barrios, C.; Saji, S.; Bondarenko, I.; Nowecki, Z.; Lian, Q.; et al. Ipatasertib plus paclitaxel for PIK3CA/AKT1/PTEN-altered hormone receptor-positive HER2-negative advanced breast cancer: Primary results from cohort B of the IPATunity130 randomized phase 3 trial. Breast Cancer Res. Treat. 2022, 191, 565–576. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Passamonti, F.; Burbury, K.; El-Galaly, T.C.; Gerds, A.; Gupta, V.; Higgins, B.; Wonde, K.; Jamois, C.; Kovic, B.; et al. The MDM2 antagonist idasanutlin in patients with polycythemia vera: Results from a single-arm phase 2 study. Blood Adv. 2022, 6, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Zhao, J.-X.; Liu, H.; Lv, J.; Yang, X.-J. Wortmannin enhances cisplatin-induced apoptosis in human ovarian cancer cells in vitro. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2428–2434. [Google Scholar]
- Boehle, A.S.; Kurdow, R.; Boenicke, L.; Schniewind, B.; Faendrich, F.; Dohrmann, P.; Kalthoff, H. Wortmannin inhibits growth of human non-small-cell lung cancer in vitro and in vivo. Langenbecks Arch. Surg. 2002, 387, 234–239. [Google Scholar] [CrossRef]
- Li, J.; Li, F.; Wang, H.; Wang, X.; Jiang, Y.; Li, D. Wortmannin reduces metastasis and angiogenesis of human breast cancer cells via nuclear factor-κB-dependent matrix metalloproteinase-9 and interleukin-8 pathways. J. Int. Med. Res. 2012, 40, 867–876. [Google Scholar] [CrossRef]
- Ghareghomi, S.; Ahmadian, S.; Zarghami, N.; Hemmati, S. hTERT-molecular targeted therapy of ovarian cancer cells via folate-functionalized PLGA nanoparticles co-loaded with MNPs/siRNA/wortmannin. Life Sci. 2021, 277, 119621. [Google Scholar] [CrossRef]
- Liu, B.; Fang, X.; Kwong, D.L.-W.; Zhang, Y.; Verhoeft, K.; Gong, L.; Zhang, B.; Chen, J.; Yu, Q.; Luo, J.; et al. Targeting TROY-mediated P85a/AKT/TBX3 signaling attenuates tumor stemness and elevates treatment response in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 182. [Google Scholar] [CrossRef] [PubMed]
- Teranishi, F.; Takahashi, N.; Gao, N.; Akamo, Y.; Takeyama, H.; Manabe, T.; Okamoto, T. Phosphoinositide 3-kinase inhibitor (wortmannin) inhibits pancreatic cancer cell motility and migration induced by hyaluronan in vitro and peritoneal metastasis in vivo. Cancer Sci. 2009, 100, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- To, A.; Medina, L.O.; Mfuh, K.O.; Lieberman, M.M.; Wong, T.A.S.; Namekar, M.; Nakano, E.; Lai, C.-Y.; Kumar, M.; Nerurkar, V.R.; et al. Recombinant Zika Virus Subunits Are Immunogenic and Efficacious in Mice. mSphere 2018, 3, e00576-17. [Google Scholar] [CrossRef]
- Crane, A.T.; Chrostek, M.R.; Krishna, V.D.; Shiao, M.; Toman, N.G.; Pearce, C.M.; Tran, S.K.; Sipe, C.J.; Guo, W.; Voth, J.P.; et al. Zika virus-based immunotherapy enhances long-term survival of rodents with brain tumors through upregulation of memory T-cells. PLoS ONE 2020, 15, e0232858. [Google Scholar] [CrossRef]
- Ávila-Pérez, G.; Nogales, A.; Park, J.-G.; Vasquez, D.M.; Dean, D.A.; Barravecchia, M.; Perez, D.R.; Almazán, F.; Martínez-Sobrido, L. In vivo rescue of recombinant Zika virus from an infectious cDNA clone and its implications in vaccine development. Sci. Rep. 2020, 10, 512. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glioblastoma (GS) Sample ID * | Responder/Non-Responder | New/Recurrent | Ethnicity | Sex (M/F) | Age | EGFR CNV | PTEN CNV | PTEN MUT |
|---|---|---|---|---|---|---|---|---|
| GS054 | Non-Responder | Recurrent | White | F | 59 | Shallow Deletion | G44D | |
| GS122 ** | Non-Responder | Recurrent | White | M | 40 | Amplified | Shallow Deletion | |
| GS147 *** | Non-Responder | Recurrent | White | M | 59 | Amplified | Shallow Deletion | |
| GS154 | Non-Responder | Recurrent | Latino | M | 51 | Copy Number Gain | ||
| GS194 + | Non-Responder | Recurrent | White | M | 52 | Amplified | Shallow Deletion | |
| GS023 | Responder | New | Mexican | F | 47 | X342_splice | ||
| GS025 | Responder | Recurrent | White | F | 39 | Amplified | ||
| GS118 + | Responder | Recurrent | White | M | 74 | Shallow Deletion | ||
| GS152 & | Responder | New | Black | M | 61 | Shallow Deletion | N292Kfs*6 | |
| GS130 $ | Responder | Recurrent | White | F | 61 | Shallow Deletion | X70_splice |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, G., Jr.; Chakravarty, N.; Paiola, S.; Urena, E.; Gyani, P.; Tse, C.; French, S.W.; Danielpour, M.; Breunig, J.J.; Nathanson, D.A.; et al. Differential Susceptibility of Ex Vivo Primary Glioblastoma Tumors to Oncolytic Effect of Modified Zika Virus. Cells 2023, 12, 2384. https://doi.org/10.3390/cells12192384
Garcia G Jr., Chakravarty N, Paiola S, Urena E, Gyani P, Tse C, French SW, Danielpour M, Breunig JJ, Nathanson DA, et al. Differential Susceptibility of Ex Vivo Primary Glioblastoma Tumors to Oncolytic Effect of Modified Zika Virus. Cells. 2023; 12(19):2384. https://doi.org/10.3390/cells12192384
Chicago/Turabian StyleGarcia, Gustavo, Jr., Nikhil Chakravarty, Sophia Paiola, Estrella Urena, Priya Gyani, Christopher Tse, Samuel W. French, Moise Danielpour, Joshua J. Breunig, David A. Nathanson, and et al. 2023. "Differential Susceptibility of Ex Vivo Primary Glioblastoma Tumors to Oncolytic Effect of Modified Zika Virus" Cells 12, no. 19: 2384. https://doi.org/10.3390/cells12192384