1. Introduction
Motor neurons (MNs) are highly polarized cells whose most extended projection, the motor axon, extends to over one meter long in adult humans. Due to the remote distances from their cell bodies (soma), distal axons and synaptic terminals depend on two distinct processes: (i) bidirectional axonal transport, facilitated by motor proteins [
1,
2] and (ii) local mRNA translation [
3,
4,
5]. The combination of these two processes enables MNs to respond efficiently to intracellular and extracellular cues crucial for their survival and function [
6]. To perform local translation, MNs must shuttle mRNAs into axons [
7,
8]. This occurs largely via RNA-binding proteins that bind specific regulatory motifs in the mRNA untranslated regions (UTRs) and also tether motor proteins [
7,
8,
9]. Previous studies found that mRNA transcripts localized to axons tend to be longer than those in the soma compartment, thus suggesting the possible role of these transcripts in RNA localization and regulation [
10,
11,
12]. Axonal and synaptic localization of micro RNAs (miRNA), as well as the RNA-induced silencing complex (RISC), was previously reported in several studies [
13,
14,
15,
16]. This implies that RNA silencing could be a mode of local regulation over mRNA translation in axons and synapses. However, knowledge is still limited regarding mechanisms of translational regulation of axonal mRNAs, and specifically of the means by which miRNA regulate their targets in axons.
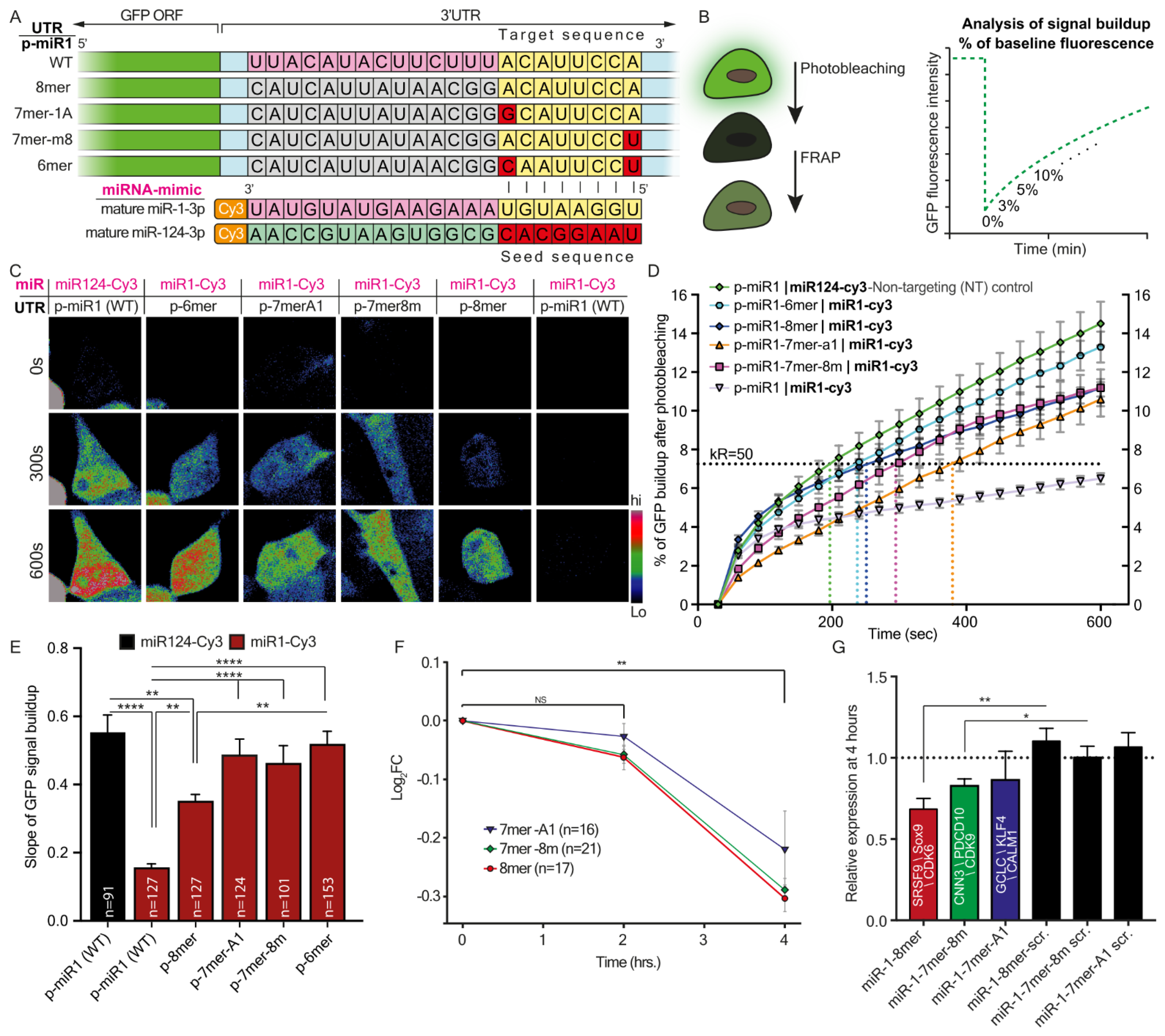
MiRNAs are small, endogenous non-coding RNAs, about 21 nucleotides long, which induce posttranscriptional silencing of their target genes, thereby functioning as negative gene expression regulators [
17]. miRNAs play a key role in regulating the transcriptome and proteome in distinct subcellular regions, including axons. In mammals, miRNAs are predicted to control the activity of about two-thirds of all coding genes [
18,
19,
20]. To exert their functional regulation, the 5′ positioned 2-8 nucleotides of the miRNA (also known as the seed region) bind the target mRNA 3′ untranslated region (3′ UTR) sequence [
17,
20,
21]. Consistent with the relatively short complementary sequence required for target gene recognition, each miRNA is estimated to target several tens to hundreds of transcripts [
22]. Analysis of conserved miRNA-3′UTR pairing has led to the identification of effective canonical site types: 8mer (bases 1–8), 7mer-m8 (bases 2–8), 7mer-A1 (bases 1–7), and 6mer (bases 2–7). Longer seed regions are believed to have greater mRNA repression efficacy [
23,
24,
25]. These canonical seeds are estimated to represent at least two-thirds of all miRNA targets. Moreover, the number of canonical sites influences target inhibition efficacy; each additional site enhances target gene repression [
26]. However, how these traits directly influence protein synthesis of target transcripts, and how this mode of regulation translates into function specifically in subcellular domains such as the neuronal axons, remain mostly unknown.
Here, using a unique live cell imaging approach, we characterized the regulatory influence of variations in miRNA:mRNA seed matching on target mRNA levels and on the mRNA translation rate. We revealed that the rate of protein synthesis is decreased in correlation to target complementation. Furthermore, we demonstrated a key mechanism in the regulation of axonal transcriptome, by which multiple repeats of miRNA target sites in the long 3′UTR of axonal mRNA variants, rather than variations in seed matching length, dominate the translation of transcripts.
2. Materials and Methods
2.1. Animals
HB9-GFP (Jax stock no. 005029) mice were originally obtained from Jackson Laboratories. The colony was maintained by breeding with ICR mice (Institute of Animal Science, Harlan Biotech, Rehovot, Israel). All the animal experiments were approved and supervised by the Animal Ethics Committee of Tel-Aviv University (Ramat-Aviv, Tel-Aviv, Israel).
2.2. Cell Culture
Monolayer-adherent HEK-293T cells (transformed human embryonic kidney cells) or SH-S5Y5 (human neuroblastoma cells) were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA), 0.3 g/liter L-glutamine, 100 units/mL penicillin, and 100 units/mL streptomycin (Biological Industries, Israel). Cells were purchased from the American Type Culture Collection (ATCC).
Cells were incubated at 37 °C in a 5% CO2 atmosphere. Before use, each cell line was confirmed to have no mycoplasma contamination using the EZ-PCR Mycoplasma Test Kit (Biological Industries, Beit-Ha’Emek, Israel). Prior to each experiment, the cells were stained with trypan blue and counted using the Countess automated cell counter (Thermo Fisher Scientific, Waltham, MA, USA).
2.3. Preparation of the Microfluidic Chamber (MFC)
Highly pure axonal RNA was isolated by using a novel radial microfluidic device fabricated according to the SU-8 photoresist protocol in the Tel-Aviv University Nano and Micro Fabrication Center. Polydimethylsiloxane (PDMS) mold was pre-treated with chlorotrimethylsilane (Merck-Sigma-Aldrich, St. Louis, MO, USA) prior to PDMS casting. PDMS base and curing agent were mixed in a ratio of 10:1. PDMS solution was mixed until it became homogenous, cast into radial microfluid chamber wafers, incubated for 2 h within a vacuum desiccator to discard trapped air bubbles, and finally baked for 3 h at 70 °C. Radial microfluidic chambers (MFCs) were then punched twice to form MFC rings. The inner well was punched with a 7 mm biopsy punch, and the outer well with a 9 mm punch. The cleaning procedure was done in a similar manner as for regular MFC. Radial MFC rings adhered to sterile 13 mm coverslips inside 24-well plates.
2.4. Primary Motor Neuron Cultures
Ventral spinal cords from E12.5 embryos were dissected in Hanks’ Balanced Salt Solution (HBSS) prior to dissociation. Dissociated MN cultures were obtained by trypsinization and trituration of explants. The upernatant was collected and centrifuged through bovine serum albumin (BSA) (Sigma) cushion. The pellet was then resuspended and centrifuged through an Optiprep (Sigma) gradient (containing 10.4% Optiprep, 10 mM Tricine, 4% w/v glucose). An MN-enriched fraction was collected from the interphase and centrifuged through a BSA cushion. The MN pellet was resuspended and plated in the central well of the radial MFC at a concentration of 250,000 MNs per chamber in 40 µL of complete neurobasal (CNB) medium, containing neurobasal, 4% B27, 2% horse serum (Biological Industries, Beit-Ha’Emek, Israel), 1% Glutamax, 1% P/S, 25 µM beta-mercapto ethanol, 25 ng/mL BDNF, 1 ng/mL GDNF (Alomone, Jerusalem, Israel), and 0.5 ng/mL CNTF (Alomone). MNs were maintained in CNB medium, which was refreshed every other day. Glial cell proliferation was restricted by the addition of 1 µM cytosine arabinoside (ARA-C; Sigma) to the culture medium in 1-3DIV. At 3DIV, the BDNF concentration in the proximal compartment was reduced (1 ng/mL), while the medium in the peripheral (axonal) compartment was enriched with GDNF and BDNF (25 ng/mL) to direct axonal growth.
2.5. RNA Extraction from MN Axons and Cell Bodies
MN axonal RNA was extracted from the outer compartment of radial MFCs at 14DIV. Axonal RNA was extracted by removing the PBS (from the prior wash) from the outer compartment and adding 100 µL Qiazol lysis reagent (Merck-Sigma–Aldrich, St. Louis, MO, USA). The inner well was filled with a higher volume of PBS to disable the inward flow of the lysis reagent toward the inner (soma) compartment and to prevent soma contamination. Axons were washed off the plate by pipetting the Qiazol reagent around the outer well for 30 s. RNA from somata in the inner compartment was extracted with 100 µL Qiazol reagent, and lysate was collected in a similar manner. RNA was isolated and purified using miRNeasy micro kit (Qiagen).
2.6. Plasmid Construction
Destabilized GFP was generated as described by Li et al. [
27]. To insert the 3′UTR counting full complementation miR-1-3p binding site, we used the QuikChange Lightning site-directed mutagenesis (SDM) kit (Agilent, Santa Clara, CA, USA). This kit was also used to create various miRNA canonical sites. The new vectors were verified by Sanger sequencing. Canonical site sequences are listed in
Table 1.
2.7. Mature miRNA Mimics
Mature miRNAs were ordered from IDT (Integrated Device Technology, San Jose, CA, USA), according to the miRBase, a miRNA database [
28].
All the miRNAs were conjugated to CY3 fluorescent dye at the 3′ end.
Mature miRNA sequences are listed in
Table 1.
2.8. Plasmid Transfection
HEK-293T cells were seeded in a pre-coated (0.01% poly-L-lysine for 30 min) 6 mm PDMS, and well adhered to a 35 mm glass-bottom dish (API) at a concentration of 8 × 103 cells/well. Twenty-four hours later, at ~60% confluence, the cells were transfected with 35 ng plasmid, or co-transfected with 33 ng plasmid and 1ng mature miRNA, using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instructions. S5Y5 cells were seeded in 24-well plates at a concentration of 1 × 105 cells/well. For miRNA overexpression (OE), transfection was performed with 500 ng of miRVec containing the desired pre-miRNA or a control vector.
2.9. qRT-PCR Analysis
Extracted total RNA was used as input for mRNA complementary deoxyribonucleic acid (cDNA) synthesis. Reverse transcription (RT) of mRNA was conducted using the random-primer and high-capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA). mRNA expression was assessed using the SYBR Green Fast PCR Master Mix (Quantabio, Beverly, MA, USA), according to the manufacturer’s instructions. miRNA expression was assessed similarly, using the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA), in accordance with the manufacturer’s instructions.
Expression values were calculated based on the comparative cycle threshold (Ct) method [
29]. mRNA expression levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an endogenous control. Specific primers for mRNA expression detection were ordered from IDT (
Table 2). GAPDH was chosen as a control due to its relatively stable expression [
30]. miRNA expression was normalized to miR-124a-3p or U6 snRNA expression (
Table 3). PCR amplification and reading were conducted on the StepOnePlus Real-Time PCR Systems (Thermo Fisher Scientific, Waltham, MA, USA).
2.10. Live Cell Confocal Imaging and Photobleaching
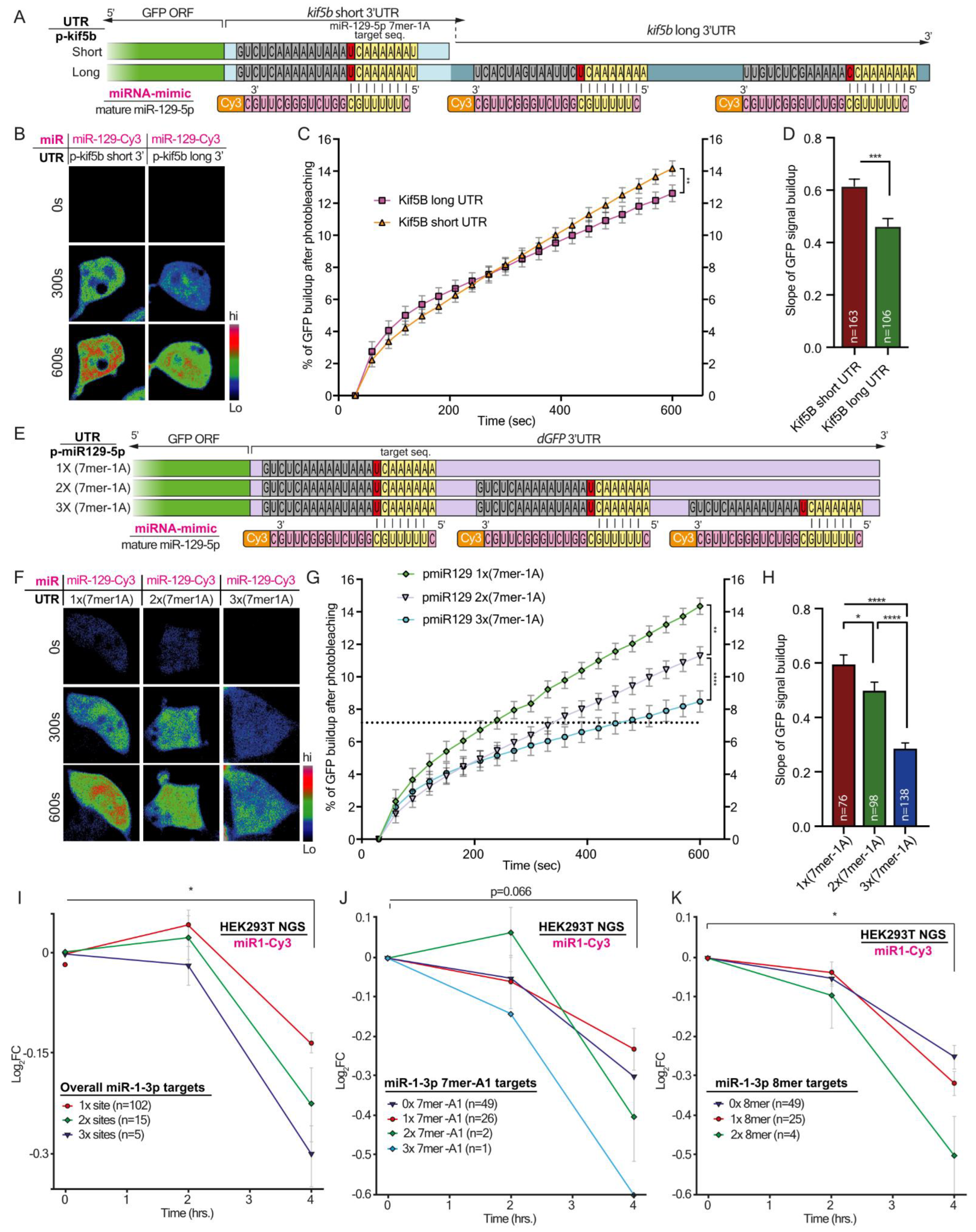
Live imaging of GFP Fluorescent Recovery After Photobleaching (FRAP) in HEK-293T cells was performed with a Nikon-Ti microscope (X40 oil objective) paired with the Andor Frappa device, Yokogawa CSU-X spinning disk, and an iXon897 EMCCD camera. FRAP on complete cells was done using a 488nm laser at 15 mW power. Dual color images of GFP and Cy3 miRNAs were acquired prior to FRAP, and for 20 consecutive times afterward at 30-s intervals. Live imaging was performed in a controlled CO2 and 37 °C chamber. Image analysis was performed with FIJI.
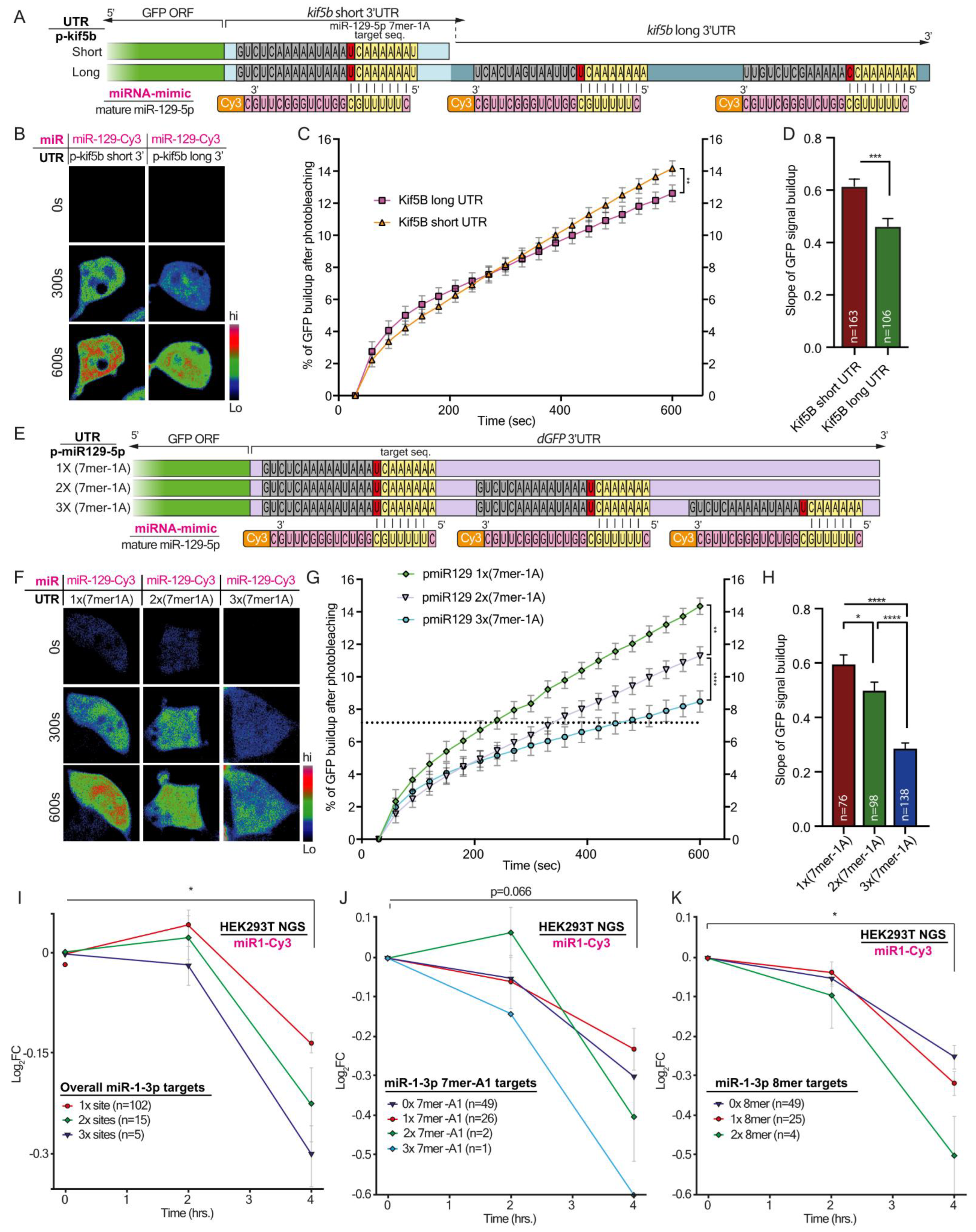
2.11. Live Cell Imaging FRAP Analysis of GFP Signal Build-up
The analysis of GFP signal build-up after photobleaching was performed with FIJI, using ‘Time series analyzer V3′ plugin. Briefly, the perimeter of the cell prior to photobleaching based on its GFP signal was marked as an ROI. The intensity of the GFP fluorescence was extracted from each time point. The signal build-up was calculated by subtracting the post-bleach fluorescence intensity value of each time point from the initial intensity, generating a normalized cumulative value for GFP build-up. The slope of dGFP signal build-up was calculated for the linear fraction of the curve (frames 5–20).
2.12. RNA Extraction
At time 0, 2, or 4 h post miR-1-3p or control RNA sequence OE, total RNA was extracted from cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), in accordance with the manufacturer’s instructions. Final RNA concentration and purity were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.13. miRNA Constructs
Pre-miRNAs were cloned into the
BamHI–EcoRI restriction site of the miRNA expression vector, miRVec, which was provided by Prof. Reuven Agami [
31]. The genomic loci of ~70 bp upstream and downstream of the pre-miRNAs were inserted into the vector.
2.14. Dual Luciferase Assay
Fragments of ~200 bp of Kif5b 3′UTR spanning the miRNA binding sites were cloned downstream to the Renilla luciferase reporter of the psiCHECK-2 plasmid (Promega, Madison, WI, USA) that contains a Firefly luciferase reporter (used as a control). The miRNA-binding sites were mutated by the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA, USA). For luciferase assays, 293T cells were transfected using Lipofectamine 2000 transfection reagent, with 5 ng psiCHECK-2 plasmid containing the desired 3′ UTR, with or without site-directed mutations; and 485 ng miRVec containing the desired pre-miRNA or a control vector. Forty-eight hours after transfection, Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System kit (Promega, Madison, WI, USA) and LUMIstar Omega Luminometer (BMG LabTech, Ortenberg, Germany), according to Promega’s instructions.
2.15. RNA-seq and Analysis
RNA quality was assessed using Agilent’s 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Samples with an RNA integrity number (RIN) >8 were considered high quality. High-quality total RNA, 1 µg, was used for ribosomal RNA (rRNA) depletion, by means of the RiboZero rRNA Removal Kit (Illumina, San Diego, CA, USA), and stranded RNA-seq libraries were constructed using a TruSeq stranded total RNA library prep kit (Illumina, San Diego, CA, USA). Then, 100 bp paired-end sequencing was performed on an Illumina Novaseq 6000 (Illumina, San Diego, CA, USA) at Macrogene corporation (Macrogene, Seoul, Republic of Korea). Library insert sizes ranged from approximately 50 to 300 bp, with an average size of 101 bp. The quality of the raw reads was assessed using FastQC software [
32]. For the DE analysis, reads were pre-processed using the ea-utils fast-mcf tool (
expressionanalysis.github.io/ea-utils/, accessed on 30 October 2019), which detects and removes adaptors and poor quality bases (Qpherd < 30) at the end of the reads [
33]. Reads were mapped to the human genome reference sequence GRCh37 (hg19) and transcriptome (GTF transcript annotation retrieved from UCSC) [
34] using the TopHat2 RNA-seq alignment tool [
35]. Gene counts were estimated using the HTSeq tool [
36].
Samples were classified as miR-1-3p or a control RNA sequence OE, with three biological replicates at each group and at each time point (0, 2, or 4 h). Normalization and differential expression were performed using the DESeq2 package in the R statistical programming environment [
37,
38].
Raw data can be accessed via the NCBI GEO repository using accession number GSE188770.
2.16. DaPars Analysis
The bioinformatics algorithm DaPars (Dynamitic analysis of Alternative Poly-Adenylation from RNA-seq) [
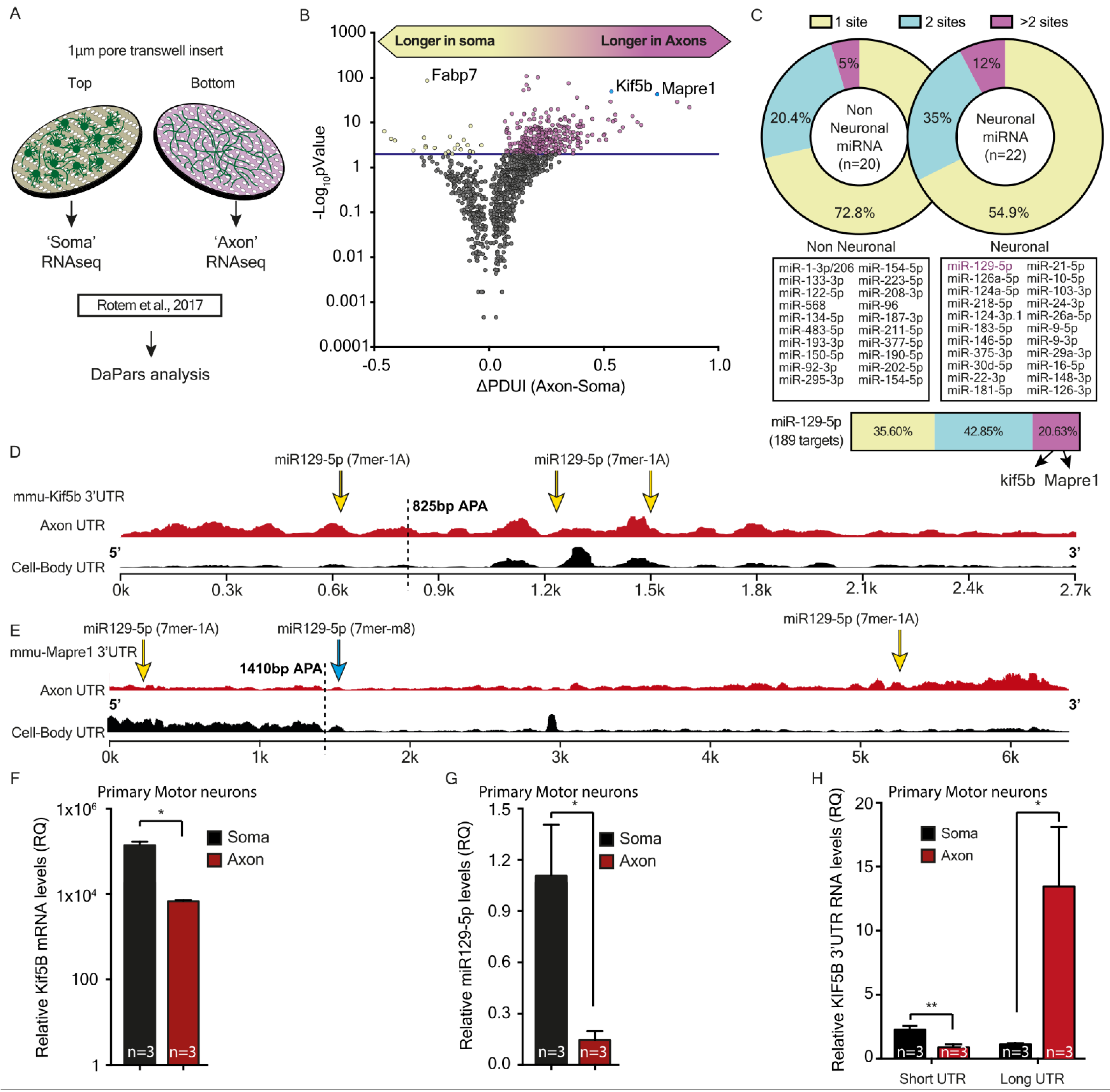
39], which uses a regression model to locate endpoints of alternative polyadenylation sites, was used to identify differences in alternative polyadenylation events between ALS samples and controls. To identify significant alternative polyadenylation (APA) events, we used the following cutoffs for the DaPars software: FDR < 0.05, |ΔPDUI| ≥ 0.2, and |dPDUI| ≥ 0.2. Accordingly, we analyzed differences in lengths of 3′UTR in mRNAs, between axons and cell-bodies.
2.17. Somatic Variant Calling
GATK4.2 was used to call short somatic variants with Mutect, using default filtering parameters to avoid low-confidence calls [
40]. Using TargetScan, we filtered only length-modifying variants that overlap 3′UTR miR binding sites.
2.18. Timespan Differential Gene Expression
Raw sequences trimmed using fastp 0.19.6 [
41] were aligned to the reference genome using STAR 2.7.3a [
42]. The raw gene expression data were processed using DESeq2 1.24.0 [
43], and miRNAs targeting differentially expressed genes were obtained from TargetScan.
2.19. Western Blot
MN-like cells (NSC-34) were cultured and transfected with either scrambled miRVec control or with miR-129-5p miRVec plasmids. Seventy-two hours after transfection, cells were lysed with a lysis solution containing PBS, 1% Triton X-100, and 1X protease inhibitor mix (Roche). Thirty µg protein lysate was loaded to SDS-PAGE and then blotted onto a nitrocellulose membrane. The membrane was blocked for 1 hour with 5% skim milk, and then incubated with mouse anti-Kif5b (kinesin heavy chain) antibody (MAB1614, Milipore; 1:500) in 5% skim milk overnight at 4 °C. The membrane was then incubated at room temperature for 1 hour with horseradish peroxidase (HRP)-anti-mouse antibody (Jackson; 1:10,000) in 5% skim milk. Rabbit anti-ERK1/2 (Abcam; 1:10,000) and HRP-anti-rabbit (Jackson; 1:10,000) were used as a loading control.
2.20. Statistics
The data are presented as means ± standard error of the mean (SEM). P-values were calculated using either ordinary ANOVA with post hoc multiple comparisons, with one/two-tail Student’s t-test, or chi-square distribution, with
p < 0.05 considered significant (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Table 4,
Table 5 and
Table 6 show descriptive endpoint statistics for the GFP-signal build-up plots presented in
Figure 1D,
Figure 2C and
Figure 3C.
4. Discussion
Axonal mRNA translation is a key process that has been shown to be critical for diverse neuronal functions, including growth and development [
9,
59,
60,
61], synaptic plasticity [
5,
62,
63,
64], response to injury [
65,
66], and local support of mitochondria [
58,
67,
68]. Previous studies, including our own, have revealed that axons and cell bodies share a large portion of their transcriptome [
7]. However, it is not completely clear how these two distinct compartments differentially regulate their local proteome. As master post-transcription regulators of gene expression, miRNAs have been repeatedly reported in axons [
17]. Nevertheless, the mechanism by which miRNA coordinates complex processes with high specificity and fidelity at the subcellular level is still not fully understood.
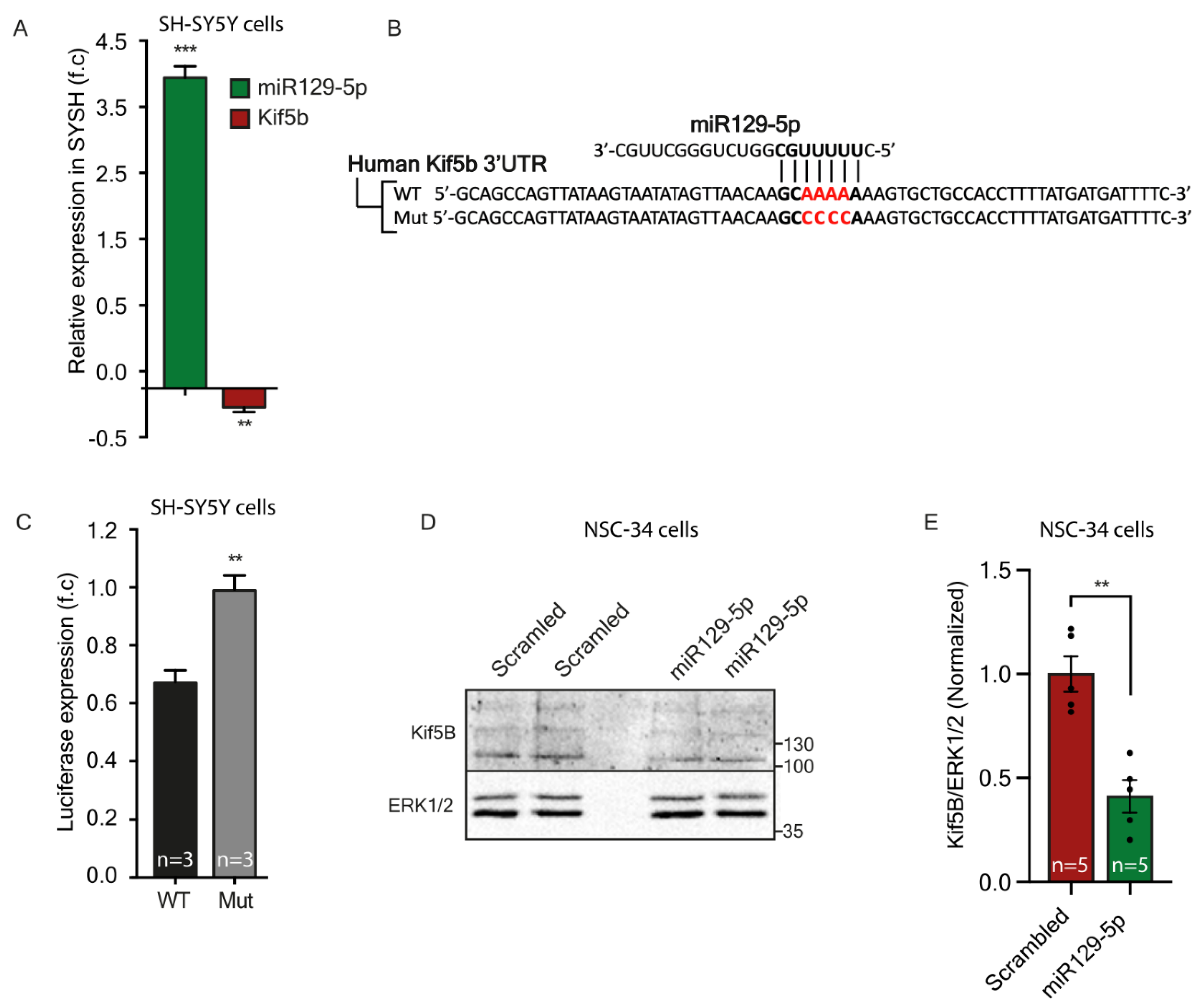
Here, by using a microscopy-based method to monitor protein synthesis rates in real-time, we revealed how modifications in the binding properties of miRNA to their target UTRs can differentially regulate transcript levels, and consequently the rate of miRNA translation. This sharpens the notion that miRNAs do not function according to the ‘all or none’ principle, but rather have a fine-tuning mode that enables dynamic control over the stability and translation of their targets. This fine-tuning mode is regulated via the number of miRNA binding sites and the type of target site. This complex level of combinatorial regulation can potentially dictate the required stoichiometry of a set of proteins for performing a specific process or function at a specific time point. Comparing NGS data from primary MNs, we found that axonal 3′UTRs of hundreds of transcripts were longer than the 3′UTRs of their corresponding transcripts in cell bodies. We showed that these long 3′UTRs contain more binding site copies for neuronal miRNAs. Specifically, miR-129-5p is predicted to target almost 40% of the transcripts with significantly longer UTRs in axons, and 17% of those with more than one binding site. We validated our findings on Kif5b, a predicted target of miR-129-5p, with three binding sites in the 3′UTR of its axonal variant, and with one of the transcripts with the longest 3′UTR in axons compared to cell bodies. We showed that in MN axons, the long 3′UTR variant of Kif5b is more abundant than the short 3′UTR variant, and contains all three binding sites for miR-129-5p. In contrast, cell bodies contain more of the short variant, with only a single miR-129-5p site. These findings imply that the long 3′UTR variant is promoted into axons, where it might also be stabilized through binding of various RNA-binding proteins, specifically to the unique 3′UTR regions. Lastly, after verifying that kif5b is a valid target of miR-129-5p, we demonstrated that the length of its UTR can determine its translation rate. This, we showed, is dependent on the number of miR-129-5p binding sites.
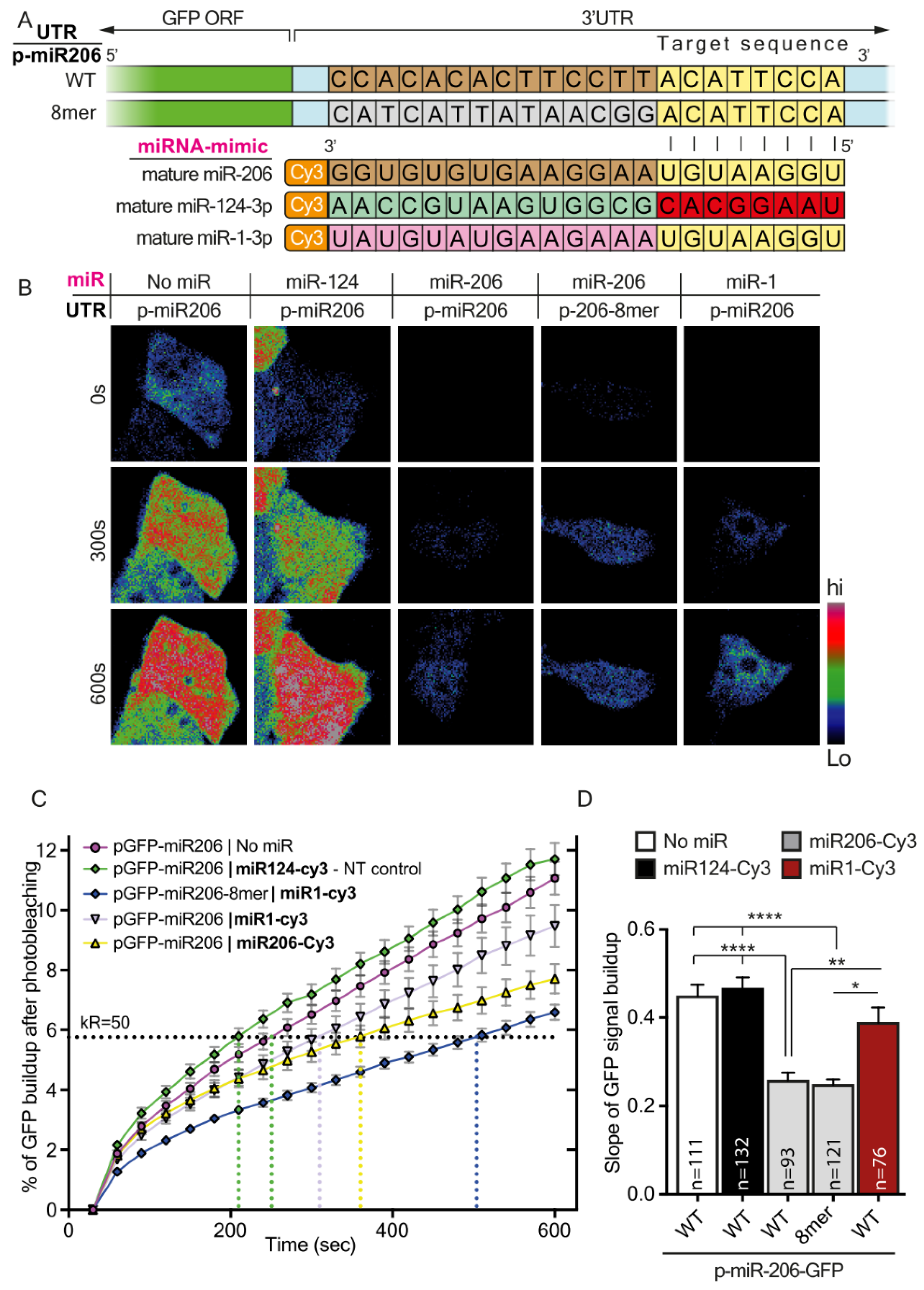
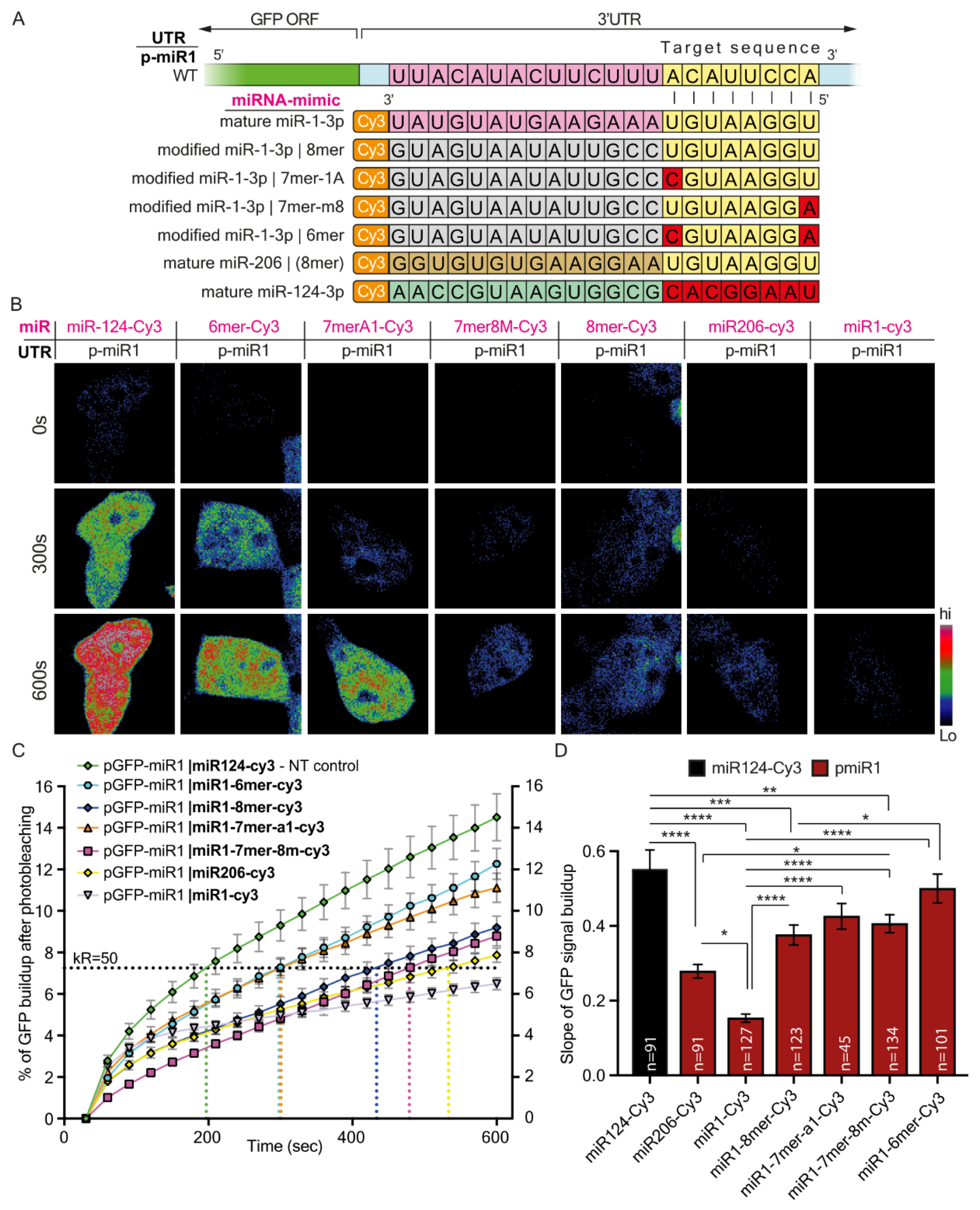
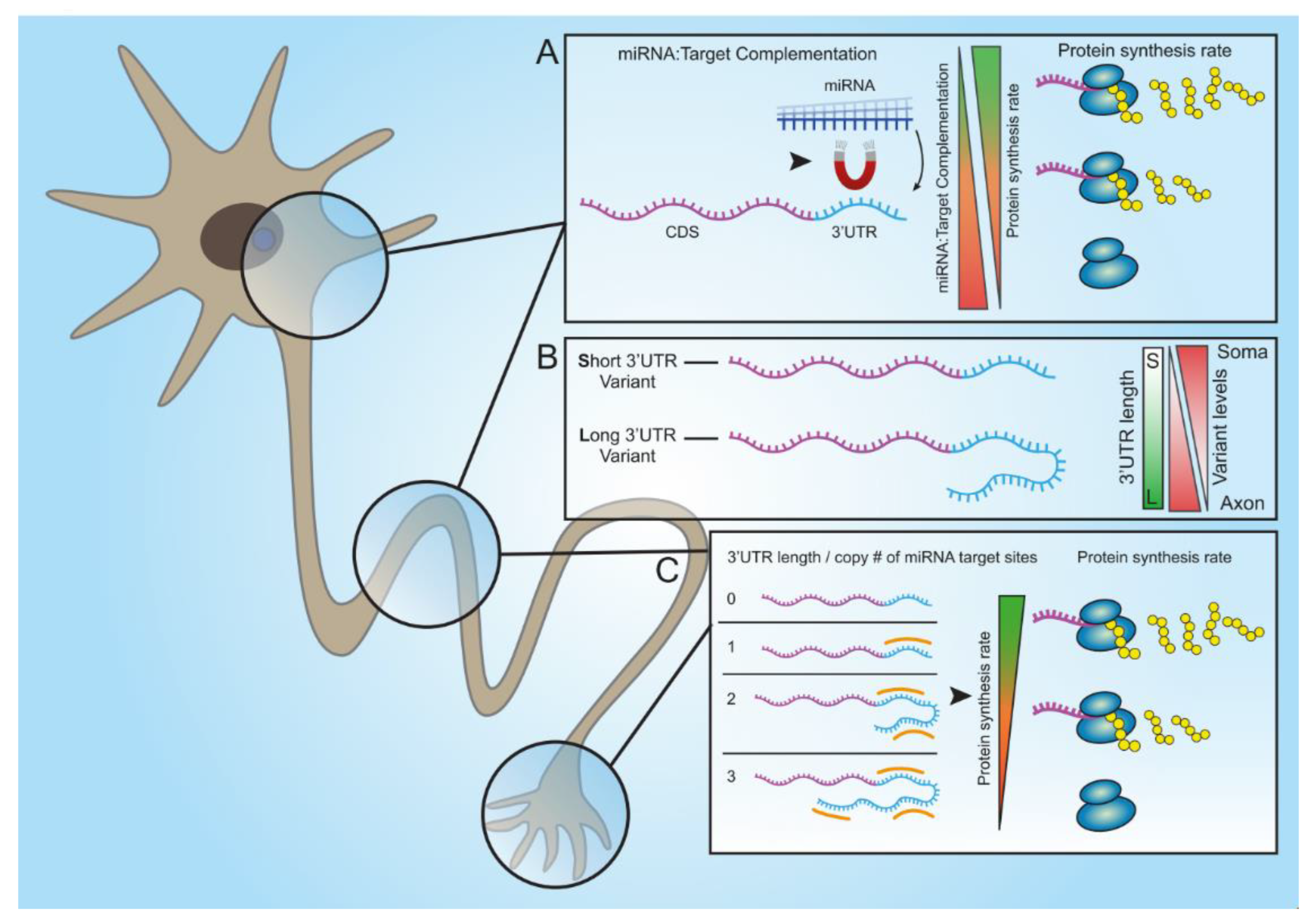
Taken together, our findings describe two mechanisms by which regulating gene expression by miRNAs is finely-tuned due to the various properties of their cognate 3′UTRs. Namely, seed-target sequence complementation, variations in the number of binding sites, or both, can determine the silencing efficacy of a single miRNA. The first mechanism seems to vary more between genes. However, the second mechanism may also apply within variants of the same gene, and thus achieve discrete regulation of subcellular transcriptomes, which is fundamental for highly polarized cells such as MNs.
In addition, for the purpose of reciprocating target complementation experiments, we designed modified miRNA mimetics. Using these, we demonstrated that the ability to fine-tune synthesis rates of a specific protein is not limited to the target sequence within the protein’s 3′UTR but can also be affected by modifying the synthetic miRNA itself.
Our findings indicate that in axons, the long 3′UTR variants are more abundant than the short ones, which may contradict the hypothesis that long 3′UTR sensitizes the transcript for stronger degradation. However, our observations were taken at a single timepoint during the development of MNs. Thus, we cannot rule out the possibility that during this time, Kif5b mRNA in axons was bound and sequestered by axonal RNA-binding proteins [
10], and that its silencing by miR-129-5p could occur only after its release for translation. Additionally, levels of miRNAs can change dramatically along the course of cellular differentiation and maturation, and also following uptake from neighboring cells. Hence, the spatiotemporal expression of miRNA-129-5p can be critical for controlling various processes at later phases of MN maturation and in a subcellular manner (e.g., synaptogenesis). Moreover, axons and synapses are strictly reliant on the spatial and temporal proteome, and unregulated synthesis and accumulation of proteins within axons and synapses could have a direct impact on their function and health. The combination of spatially localized axonal miRNAs together with more susceptible mRNAs, can perhaps enable the spatiotemporal distribution of mRNAs across the axonal sub-compartments, as well as restrict the random spreading of transcripts throughout the cell.
Aberrant miRNA expression [
7,
69,
70] and deficits in RNA processing and translation [
4,
68,
71], and also defective axonal transport [
2,
72,
73] and mitochondrial abnormalities [
74], are mechanisms associated with MN degeneration in amyotrophic lateral sclerosis (ALS). Interestingly, several recent works have specifically identified miR-129-5p as a new ALS-linked miRNA and suggest it confers protective traits [
75,
76]. Intriguingly, our unbiased approach identified miR-129-5p as a potential master regulator of axonal proteome and highlights the motor protein Kif5b and the microtubule-associated Mapre1 as two of its ‘multiple binding site’ targets in axons. Kif5b mediates the anterograde axonal transport of diverse organelles, including mitochondria, into distal axons and synapses [
56,
57,
77]. Recently, the transcripts of several kinesin superfamily motor proteins were identified in distal axons and presynaptic terminals of excitatory neurons. This suggests that these are locally translated to efficiently transport RNA granules, newly synthesized cargo, and mitochondria into the synapse [
5]. Specifically, mutations in the Kif5A gene, a closely related kinesin, are causes of various neurological disorders including Charcot-Marie Tooth and ALS [
73,
78,
79]. Therefore, our findings potentially link the critical deficits in ALS and suggest that miRNA-129-5p may regulate the axonal local synthesis of Kif5b, which consequently participates in the vital process of axonal transport. Future research should investigate the specific involvement of miR-129-5p in ALS pathogenesis, and attempt to unveil its axonal targets and the local processes it regulates.
To summarize, our findings illustrate several mechanisms that cells have adapted to achieve an efficient, versatile, yet potent response to the expression of a single miRNA. These involve changes in target complementation and the number of miRNA binding sites; and distinctly distributing transcript variants to subcellular compartments.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}