

Milk Fat Globule Epidermal Growth Factor VIII Fragment Medin in Age-Associated Arterial Adverse Remodeling and Arterial Disease

Abstract
:1. Introduction
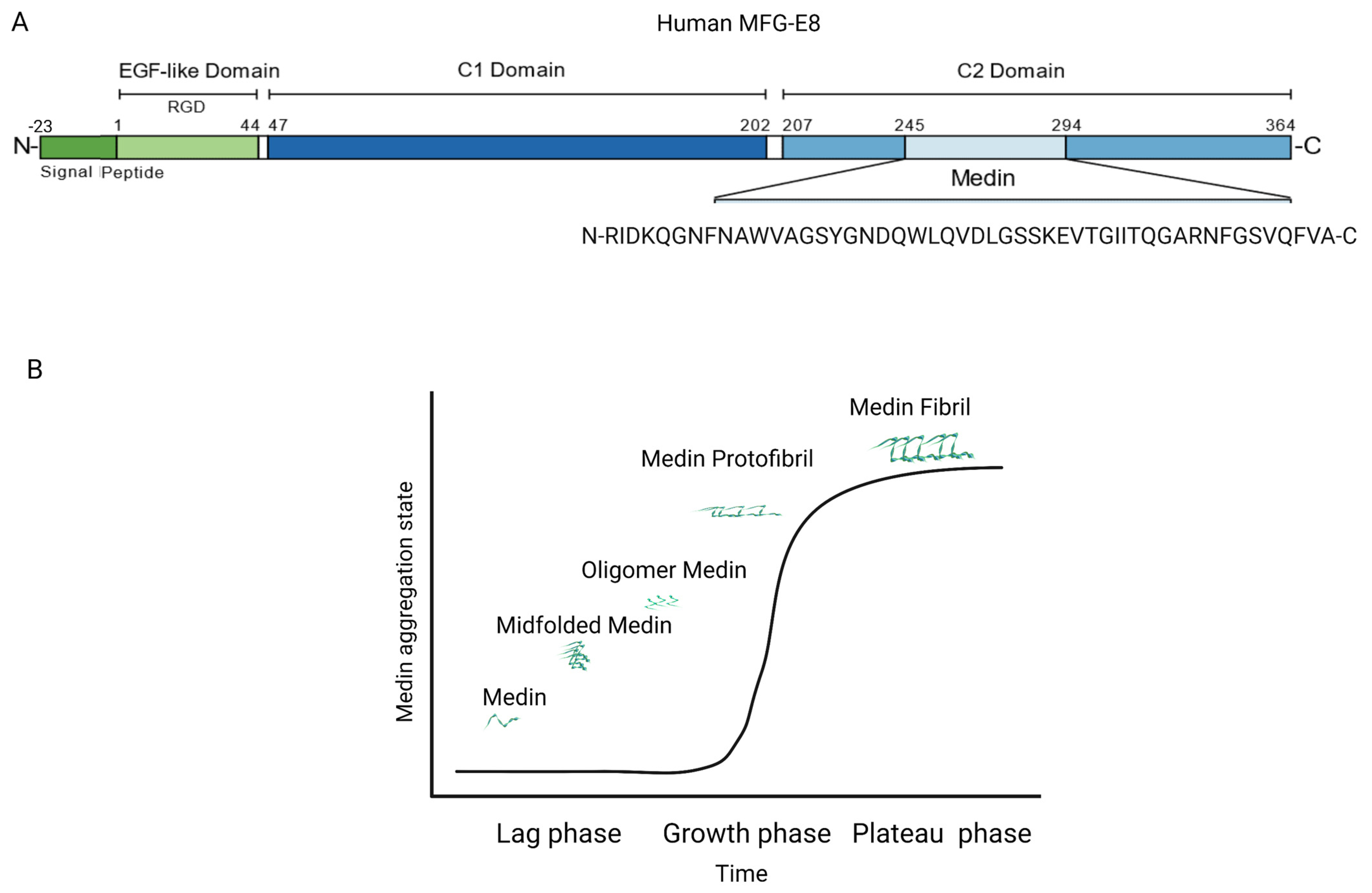
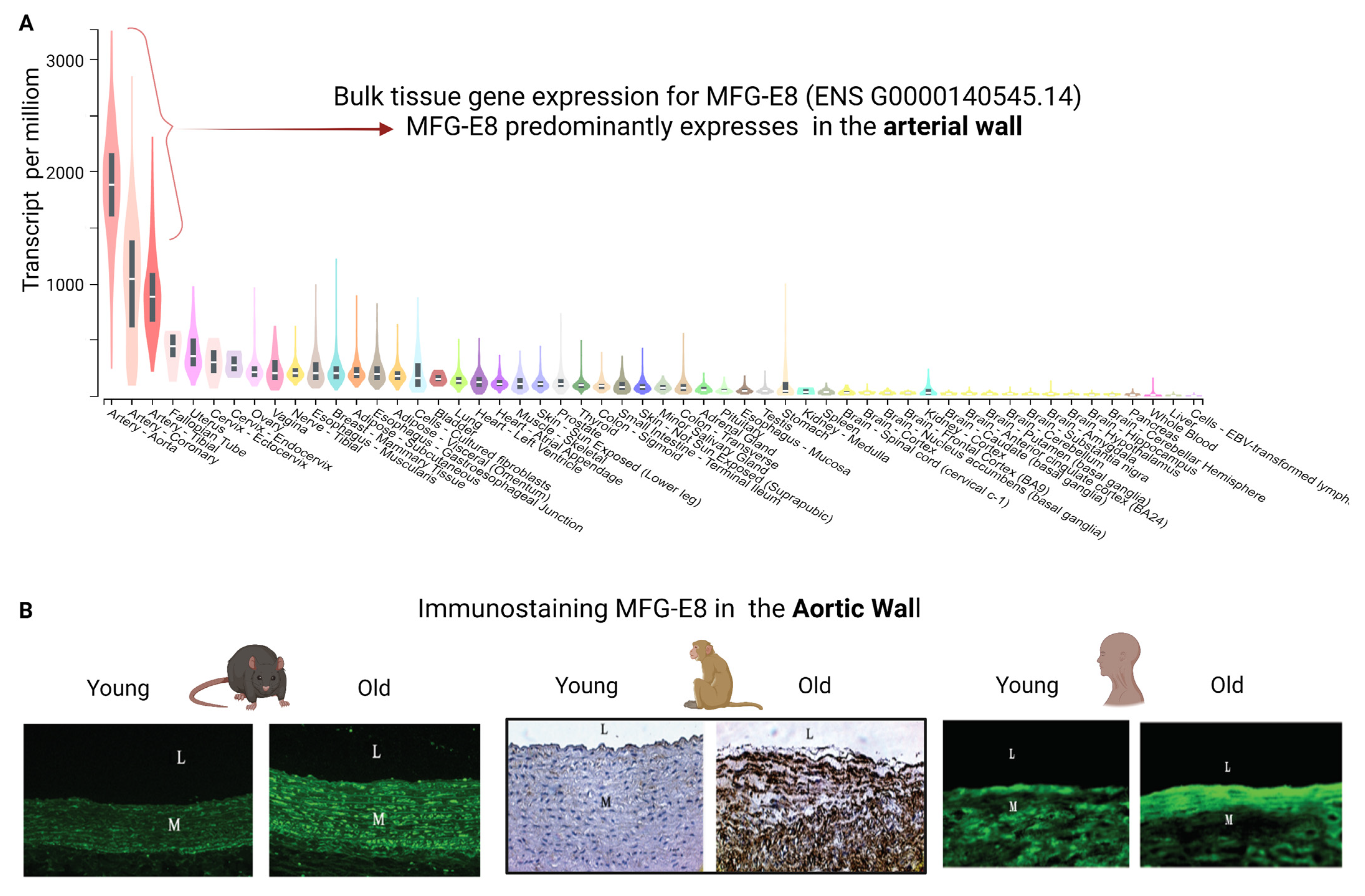
2. MFG-E8 and Its Fragment Medin Expression during Age-Associated Inflammatory Remodeling
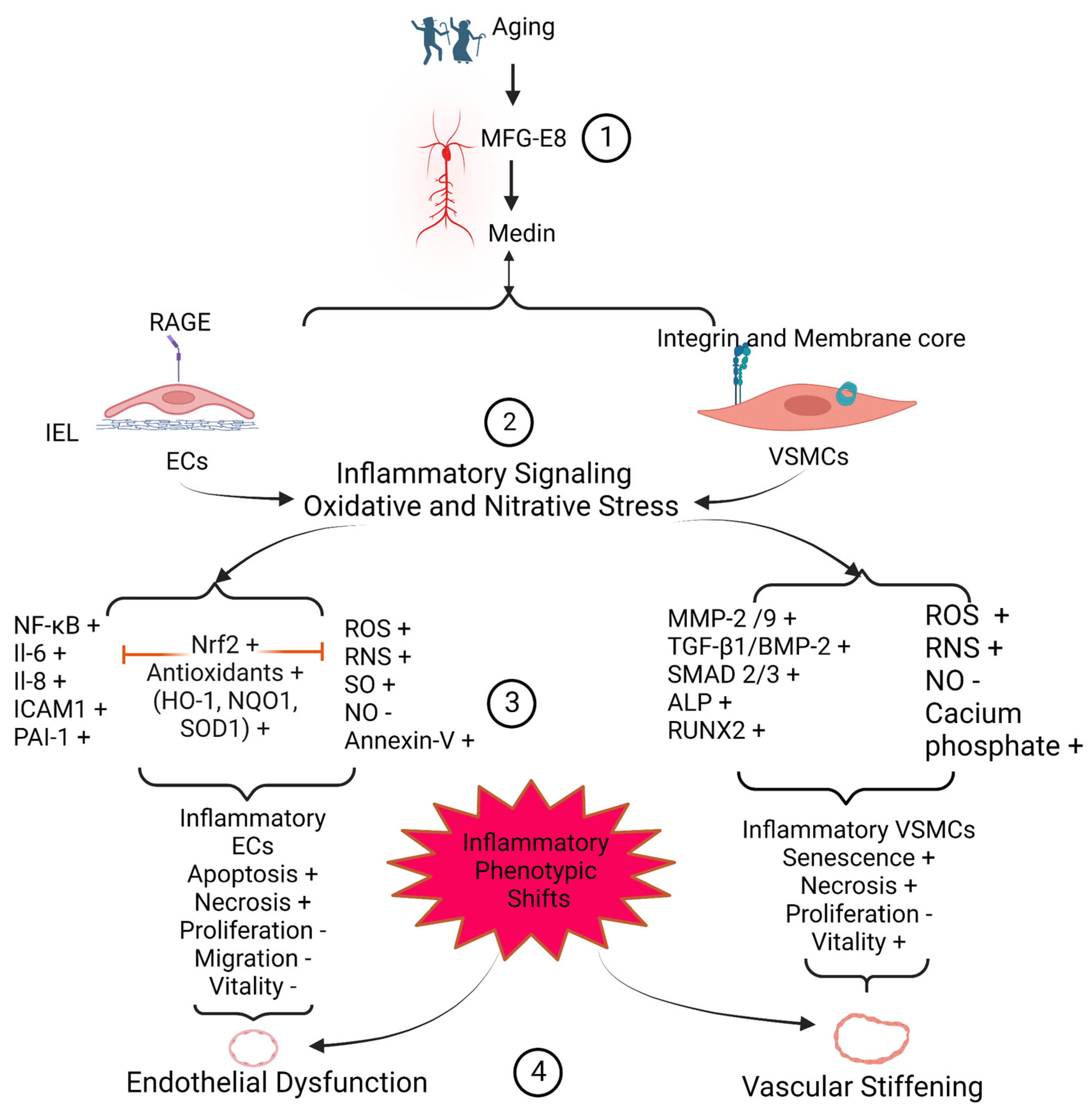
3. Medin Induces Vascular Cell Inflammation
3.1. EC Inflammation and Viability
3.2. VSMC Inflammation and Viability
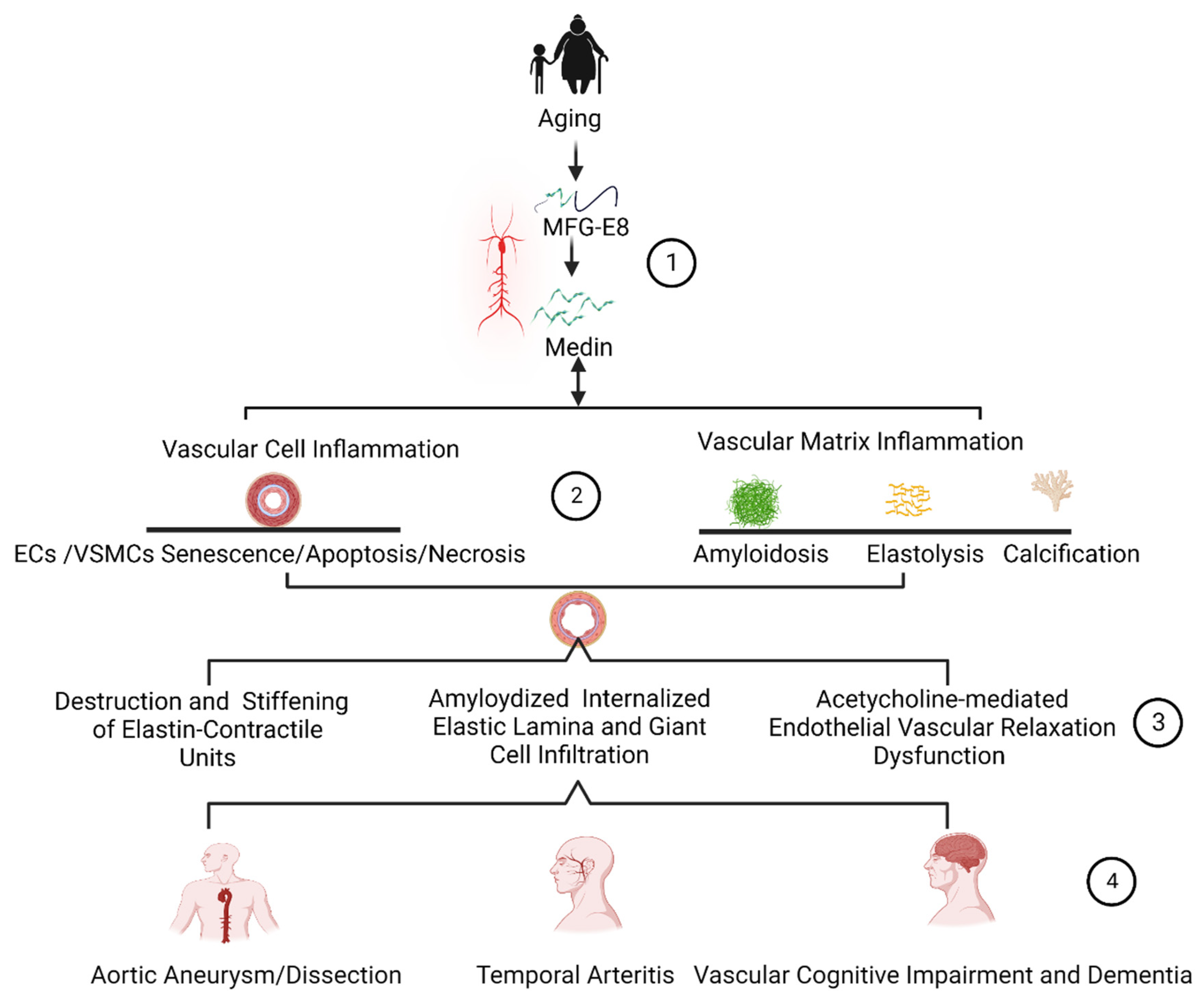
4. Medin Remodels Extracellular Matrices
4.1. Amyloidosis
4.2. Elastolysis
4.3. Calcification
5. Medin Deposition Impairs Vascular Function
5.1. Arterial Endothelial Disorder and Dysfunction
5.2. Arterial Microstructure Disorder and Stiffening
6. Medin Deposition Is Involved in the Pathogenesis of Vascular Diseases
6.1. Arterial Aneurysms and Dissections
6.2. Temporal Arteritis
6.3. Vascular-Related Cognitive Impairment and Dementia
7. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Degenhardt, K.; Wagner, J.; Skodras, A.; Candlish, M.; Koppelmann, A.J.; Wild, K.; Maxwell, R.; Rotermund, C.; von Zweydorf, F.; Gloeckner, C.J.; et al. Medin aggregation causes cerebrovascular dysfunction in aging wild-type mice. Proc. Natl. Acad. Sci. USA 2020, 117, 23925–23931. [Google Scholar] [CrossRef]
- Karamanova, N.; Truran, S.; Serrano, G.E.; Beach, T.G.; Madine, J.; Weissig, V.; Davies, H.A.; Veldhuizen, J.; Nikkhah, M.; Hansen, M.; et al. Endothelial Immune Activation by Medin: Potential Role in Cerebrovascular Disease and Reversal by Monosialoganglioside-Containing Nanoliposomes. J. Am. Heart Assoc. 2020, 9, e014810. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, L.; Monticone, R.E.; Lakatta, E.G. Proinflammation: The key to arterial aging. Trends Endocrinol. Metab. 2014, 25, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Migrino, R.Q.; Karamanova, N.; Truran, S.; Serrano, G.E.; Davies, H.A.; Madine, J.; Beach, T.G. Cerebrovascular medin is associated with Alzheimer’s disease and vascular dementia. Alzheimer’s Dement. 2020, 12, e12072. [Google Scholar] [CrossRef]
- Kim, S.H.; Monticone, R.E.; McGraw, K.R.; Wang, M. Age-Associated Proinflammatory Elastic Fiber Remodeling in Large Arteries. Mech. Ageing Dev. 2021, 196, 111490. [Google Scholar] [CrossRef]
- Wang, M.; Monticon, R.; McGraw, K. Proinflammation, profibrosis, and arterial aging. Aging Med. 2020, 3, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Miura, Y.; Tsumoto, H.; Iwamoto, M.; Yamaguchi, Y.; Ko, P.; Soejima, Y.; Yoshida, S.; Toda, T.; Arai, T.; Hamamatsu, A.; et al. Age-associated proteomic alterations in human aortic media. Geriatr. Gerontol. Int. 2019, 19, 1054–1062. [Google Scholar] [CrossRef]
- Watson, N.L.; Sutton-Tyrrell, K.; Rosano, C.; Boudreau, R.M.; Hardy, S.E.; Simonsick, E.M.; Najjar, S.S.; Launer, L.J.; Yaffe, K.; Atkinson, H.H.; et al. Arterial stiffness and cognitive decline in well-functioning older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 1336–1342. [Google Scholar] [CrossRef]
- Migrino, R.Q.; Truran, S.; Karamanova, N.; Serrano, G.E.; Madrigal, C.; Davies, H.A.; Madine, J.; Reaven, P.; Beach, T.G. Human cerebral collateral arteriole function in subjects with normal cognition, mild cognitive impairment, and dementia. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H284–H290. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, H.H.; Lakatta, E.G. Milk fat globule epidermal growth factor VIII signaling in arterial wall remodeling. Curr. Vasc. Pharmacol. 2013, 11, 768–776. [Google Scholar] [CrossRef]
- Westermark, G.T.; Westermark, P. Localized amyloids important in diseases outside the brain--lessons from the islets of Langerhans and the thoracic aorta. FEBS J. 2011, 278, 3918–3929. [Google Scholar] [CrossRef]
- Haggqvist, B.; Naslund, J.; Sletten, K.; Westermark, G.T.; Mucchiano, G.; Tjernberg, L.O.; Nordstedt, C.; Engstrom, U.; Westermark, P. Medin: An integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 8669–8674. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A.; Malmstrom, S.; Westermark, P. Signs of cross-seeding: Aortic medin amyloid as a trigger for protein AA deposition. Amyloid 2011, 18, 229–234. [Google Scholar] [CrossRef]
- Larsson, A.; Peng, S.; Persson, H.; Rosenbloom, J.; Abrams, W.R.; Wassberg, E.; Thelin, S.; Sletten, K.; Gerwins, P.; Westermark, P. Lactadherin binds to elastin--a starting point for medin amyloid formation? Amyloid 2006, 13, 78–85. [Google Scholar] [CrossRef]
- Larsson, A.; Soderberg, L.; Westermark, G.T.; Sletten, K.; Engstrom, U.; Tjernberg, L.O.; Naslund, J.; Westermark, P. Unwinding fibril formation of medin, the peptide of the most common form of human amyloid. Biochem. Biophys. Res. Commun. 2007, 361, 822–828. [Google Scholar] [CrossRef]
- Peng, S.; Glennert, J.; Westermark, P. Medin-amyloid: A recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 2005, 12, 96–102. [Google Scholar] [CrossRef]
- Peng, S.; Westermark, G.T.; Naslund, J.; Haggqvist, B.; Glennert, J.; Westermark, P. Medin and medin-amyloid in ageing inflamed and non-inflamed temporal arteries. J. Pathol. 2002, 196, 91–96. [Google Scholar] [CrossRef]
- Chiang, H.Y.; Chu, P.H.; Lee, T.H. MFG-E8 mediates arterial aging by promoting the proinflammatory phenotype of vascular smooth muscle cells. J. Biomed. Sci. 2019, 26, 61. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Liu, L.; Zhu, W.; Telljohann, R.; Zhang, J.; Monticone, R.E.; McGraw, K.R.; Liu, C.; Morrell, C.H.; Garrido-Gil, P.; et al. Inflammatory Role of Milk Fat Globule-Epidermal Growth Factor VIII in Age-Associated Arterial Remodeling. J. Am. Heart Assoc. 2022, 11, e022574. [Google Scholar] [CrossRef]
- Wang, M.; Fu, Z.; Wu, J.; Zhang, J.; Jiang, L.; Khazan, B.; Telljohann, R.; Zhao, M.; Krug, A.W.; Pikilidou, M. MFG-E8 activates proliferation of vascular smooth muscle cells via integrin signaling. Aging Cell 2012, 11, 500–508. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, M.; Gucek, M.; Zhang, J.; Wu, J.; Jiang, L.; Monticone, R.E.; Khazan, B.; Telljohann, R.; Mattison, J.; et al. Milk fat globule protein epidermal growth factor-8: A pivotal relay element within the angiotensin II and monocyte chemoattractant protein-1 signaling cascade mediating vascular smooth muscle cells invasion. Circ. Res. 2009, 104, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.A.; Caamano-Gutierrez, E.; Chim, Y.H.; Field, M.; Nawaytou, O.; Ressel, L.; Akhtar, R.; Madine, J. Idiopathic degenerative thoracic aneurysms are associated with increased aortic medial amyloid. Amyloid 2019, 26, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Middleton, D.A. Comparison of aggregation enhancement and inhibition as strategies for reducing the cytotoxicity of the aortic amyloid polypeptide medin. Eur. Biophys. J. 2010, 39, 1281–1288. [Google Scholar] [CrossRef]
- Peng, S.; Larsson, A.; Wassberg, E.; Gerwins, P.; Thelin, S.; Fu, X.; Westermark, P. Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab. Invest. 2007, 87, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.P.; Hsu, M.E.; Chiou, Y.Y.; Hsu, H.Y.; Tsai, H.C.; Peng, Y.J.; Lu, C.Y.; Pan, C.Y.; Yu, W.C.; Chen, C.H.; et al. Comparative proteomic analysis of rat aorta in a subtotal nephrectomy model. Proteomics 2010, 10, 2429–2443. [Google Scholar] [CrossRef]
- Ni, Y.Q.; Zhan, J.K.; Liu, Y.S. Roles and mechanisms of MFG-E8 in vascular aging-related diseases. Ageing Res. Rev. 2020, 64, 101176. [Google Scholar] [CrossRef]
- Wagner, J.; Degenhardt, K.; Veit, M.; Louros, N.; Konstantoulea, K.; Skodras, A.; Wild, K.; Liu, P.; Obermüller, U.; Bansal, V.; et al. Medin co-aggregates with vascular amyloid-β in Alzheimer’s disease. Nature 2022, 612, 123–131. [Google Scholar] [CrossRef]
- Migrino, R.Q.; Davies, H.A.; Truran, S.; Karamanova, N.; Franco, D.A.; Beach, T.G.; Serrano, G.E.; Truong, D.; Nikkhah, M.; Madine, J. Amyloidogenic medin induces endothelial dysfunction and vascular inflammation through the receptor for advanced glycation endproducts. Cardiovasc. Res. 2017, 113, 1389–1402. [Google Scholar] [CrossRef] [Green Version]
- Hinterseher, I.; Erdman, R.; Elmore, J.R.; Stahl, E.; Pahl, M.C.; Derr, K.; Golden, A.; Lillvis, J.H.; Cindric, M.C.; Jackson, K.; et al. Novel pathways in the pathobiology of human abdominal aortic aneurysms. Pathobiology 2013, 80, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Wang, M.Q.; Ni, S.H.; Wang, M.; Liu, L.Y.; You, H.Y.; Wu, X.H.; Wang, Y.J.; Lu, L.; Wei, L.B. Salidroside ameliorates endothelial inflammation and oxidative stress by regulating the AMPK/NF-kappaB/NLRP3 signaling pathway in AGEs-induced HUVECs. Eur. J. Pharmacol. 2020, 867, 172797. [Google Scholar] [CrossRef]
- Kim, S.H.; Liu, L.; Ni, L.; Zhang, L.; Zhang, J.; Wang, Y.; McGraw, K.R.; Monticone, R.; Telljohann, R.; Morrell, C.H.; et al. Effects of Milk Fat Globule Epidermal Growth Factor VIII On Age-Associated Arterial Elastolysis, Fibrosis, and Calcification. bioRxiv 2022. [Google Scholar] [CrossRef]
- Davies, H.A.; Phelan, M.M.; Wilkinson, M.C.; Migrino, R.Q.; Truran, S.; Franco, D.A.; Liu, L.N.; Longmore, C.J.; Madine, J. Oxidative Stress Alters the Morphology and Toxicity of Aortic Medial Amyloid. Biophys. J. 2015, 109, 2363–2370. [Google Scholar] [CrossRef] [Green Version]
- Karamanova, N.; Truran, S.; Weissig, V.; Madine, J.; Davies, H.; Guzman-Villanueva, D.; Migrino, R. Amyloidogenic Medin Impairs Endothelial Cell Autophagy and Viability that is Reversed by Monosialoganglioside Nanoliposomes. FASEB J. 2018, 32, 846.816. [Google Scholar] [CrossRef]
- Li, B.Y.; Li, X.L.; Cai, Q.; Gao, H.Q.; Cheng, M.; Zhang, J.H.; Wang, J.F.; Yu, F.; Zhou, R.H. Induction of lactadherin mediates the apoptosis of endothelial cells in response to advanced glycation end products and protective effects of grape seed procyanidin B2 and resveratrol. Apoptosis 2011, 16, 732–745. [Google Scholar] [CrossRef]
- Li, B.Y.; Li, X.L.; Gao, H.Q.; Zhang, J.H.; Cai, Q.; Cheng, M.; Lu, M. Grape seed procyanidin B2 inhibits advanced glycation end product-induced endothelial cell apoptosis through regulating GSK3beta phosphorylation. Cell Biol. Int. 2011, 35, 663–669. [Google Scholar] [CrossRef]
- Nerelius, C.; Gustafsson, M.; Nordling, K.; Larsson, A.; Johansson, J. Anti-amyloid activity of the C-terminal domain of proSP-C against amyloid beta-peptide and medin. Biochemistry 2009, 48, 3778–3786. [Google Scholar] [CrossRef]
- Whitehead, M.; Yusoff, S.; Ahmad, S.; Schmidt, L.; Mayr, M.; Madine, J.; Middleton, D.; Shanahan, C.M. Vascular smooth muscle cell senescence accelerates medin aggregation via small extracellular vesicle secretion and extracellular matrix reorganization. Aging Cell 2022, e13746. [Google Scholar] [CrossRef]
- Davies, H.A.; Madine, J.; Middleton, D.A. Comparisons with amyloid-beta reveal an aspartate residue that stabilizes fibrils of the aortic amyloid peptide medin. J. Biol. Chem. 2015, 290, 7791–7803. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.-Q.; Li, S.; Lin, X.; Wang, Y.-J.; He, J.-Y.; Song, W.-L.; Xiang, Q.-Y.; Zhao, Y.; Li, C.; Wang, Y.; et al. Exosomal MFGE8 from high glucose induced endothelial cells is involved in calcification/senescence of vascular smooth muscle cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Younger, S.; Jang, H.; Davies, H.A.; Niemiec, M.J.; Garcia, J.G.N.; Nussinov, R.; Migrino, R.Q.; Madine, J.; Arce, F.T. Medin Oligomer Membrane Pore Formation: A Potential Mechanism of Vascular Dysfunction. Biophys. J. 2020, 118, 2769–2782. [Google Scholar] [CrossRef]
- Davies, H.A.; Madine, J.; Middleton, D.A. Solid-state NMR reveals differences in the packing arrangements of peptide aggregates derived from the aortic amyloid polypeptide medin. J. Pept. Sci. 2012, 18, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.; Ahmad, S.; Shanahan, C. BS47 Role of vascular smooth muscle cell-derived exosomes in age-related vascular amyloidosis. Heart 2019, 105, A169–A170. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.R.; Dobson, C.M. In vitro characterization of lactoferrin aggregation and amyloid formation. Biochemistry 2003, 42, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Mucchiano, G.; Cornwell, G.G., 3rd; Westermark, P. Senile aortic amyloid. Evidence for two distinct forms of localized deposits. Am. J. Pathol. 1992, 140, 871–877. [Google Scholar]
- Davies, H.A.; Wilkinson, M.C.; Gibson, R.P.; Middleton, D.A. Expression and purification of the aortic amyloid polypeptide medin. Protein Expr. Purif. 2014, 98, 32–37. [Google Scholar] [CrossRef]
- Olofsson, A.; Borowik, T.; Grobner, G.; Sauer-Eriksson, A.E. Negatively charged phospholipid membranes induce amyloid formation of medin via an alpha-helical intermediate. J. Mol. Biol. 2007, 374, 186–194. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Amyloidogenic hexapeptide fragment of medin: Homology to functional islet amyloid polypeptide fragments. Amyloid 2004, 11, 81–89. [Google Scholar] [CrossRef]
- Boraldi, F.; Moscarelli, P.; Bochicchio, B.; Pepe, A.; Salvi, A.M.; Quaglino, D. Heparan sulfates facilitate harmless amyloidogenic fibril formation interacting with elastin-like peptides. Sci. Rep. 2018, 8, 3115. [Google Scholar] [CrossRef] [Green Version]
- Dandurand, J.; Samouillan, V.; Lacoste-Ferre, M.H.; Lacabanne, C.; Bochicchio, B.; Pepe, A. Conformational and thermal characterization of a synthetic peptidic fragment inspired from human tropoelastin: Signature of the amyloid fibers. Pathol. Biol. 2014, 62, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.-Y.; Chu, P.-H.; Chen, S.-C.; Lee, T.-H. MFG-E8 promotes osteogenic transdifferentiation of smooth muscle cells and vascular calcification by regulating TGF-β1 signaling. Commun. Biol. 2022, 5, 364. [Google Scholar] [CrossRef]
- Marie, P.; Labas, V.; Brionne, A.; Harichaux, G.; Hennequet-Antier, C.; Nys, Y.; Gautron, J. Quantitative proteomics and bioinformatic analysis provide new insight into protein function during avian eggshell biomineralization. J. Proteom. 2015, 113, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.; Labas, V.; Brionne, A.; Harichaux, G.; Hennequet-Antier, C.; Rodriguez-Navarro, A.B.; Nys, Y.; Gautron, J. Quantitative proteomics provides new insights into chicken eggshell matrix protein functions during the primary events of mineralisation and the active calcification phase. J. Proteom. 2015, 126, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Stapane, L.; Le Roy, N.; Hincke, M.T.; Gautron, J. The glycoproteins EDIL3 and MFGE8 regulate vesicle-mediated eggshell calcification in a new model for avian biomineralization. J. Biol. Chem. 2019, 294, 14526–14545. [Google Scholar] [CrossRef] [PubMed]
- Sinder, B.P.; Zweifler, L.; Koh, A.J.; Michalski, M.N.; Hofbauer, L.C.; Aguirre, J.I.; Roca, H.; McCauley, L.K. Bone Mass Is Compromised by the Chemotherapeutic Trabectedin in Association With Effects on Osteoblasts and Macrophage Efferocytosis. J. Bone Miner. Res. 2017, 32, 2116–2127. [Google Scholar] [CrossRef] [Green Version]
- Ushiki, T. Collagen fibers, reticular fibers and elastic fibers. A comprehensive understanding from a morphological viewpoint. Arch. Histol. Cytol. 2002, 65, 109–126. [Google Scholar] [CrossRef] [Green Version]
- Raspanti, M.; Protasoni, M.; Manelli, A.; Guizzardi, S.; Mantovani, V.; Sala, A. The extracellular matrix of the human aortic wall: Ultrastructural observations by FEG-SEM and by tapping-mode AFM. Micron 2006, 37, 81–86. [Google Scholar] [CrossRef]
- Berquand, A.; Wahart, A.; Henry, A.; Gorisse, L.; Maurice, P.; Blaise, S.; Romier-Crouzet, B.; Pietrement, C.; Bennasroune, A.; Sartelet, H.; et al. Revealing the elasticity of an individual aortic fiber during ageing at nanoscale by in situ atomic force microscopy. Nanoscale 2021, 13, 1124–1133. [Google Scholar] [CrossRef]
- Kuznetsova, T.G.; Starodubtseva, M.N.; Yegorenkov, N.I.; Chizhik, S.A.; Zhdanov, R.I. Atomic force microscopy probing of cell elasticity. Micron 2007, 38, 824–833. [Google Scholar] [CrossRef]
- Le Master, E.; Fancher, I.S.; Lee, J.; Levitan, I. Comparative analysis of endothelial cell and sub-endothelial cell elastic moduli in young and aged mice: Role of CD36. J. Biomech. 2018, 76, 263–268. [Google Scholar] [CrossRef]
- Jannatbabaei, A.; Tafazzoli-Shadpour, M.; Seyedjafari, E. Effects of substrate mechanics on angiogenic capacity and nitric oxide release in human endothelial cells. Ann. N. Y. Acad. Sci. 2020, 1470, 31–43. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.M.; Kucinska, M.; Jakubowska, A.; Sokolowska, P.; Rosowski, M.; Tkacz-Szczesna, B.; Komorowski, P.; Makowski, K.; Walkowiak, B. Endothelial cell aging detection by means of atomic force spectroscopy. J. Mol. Recognit. 2020, 33, e2853. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Kim, B.C.; Wang, M.; Huang, J.; Isak, A.; Bexiga, N.M.; Monticone, R.; Ha, T.; Lakatta, E.G.; An, S.S. TGFβ1 reinforces arterial aging in the vascular smooth muscle cell through a long-range regulation of the cytoskeletal stiffness. Sci. Rep. 2018, 8, 2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.D.; Arora, R.R. Pathophysiology, diagnosis, and management of aortic dissection. Ther. Adv. Cardiovasc. Dis. 2008, 2, 439–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Muniz Castro, H.M.; Bhattacharjee, M.B.; Chaudhry, I.A.; Chuang, A.Z.; Mankiewicz, K.A.; Adesina, O.O. Diagnosis of giant cell arteritis using clinical, laboratory, and histopathological findings in patients undergoing temporal artery biopsy. Clin. Neurol. Neurosurg. 2022, 221, 107377. [Google Scholar] [CrossRef]
- Sharma, A.; Mohammad, A.J.; Turesson, C. Incidence and prevalence of giant cell arteritis and polymyalgia rheumatica: A systematic literature review. Semin. Arthritis Rheum. 2020, 50, 1040–1048. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Medium- and large-vessel vasculitis. N. Engl. J. Med. 2003, 349, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Breteler, M.M.; van Harskamp, F.; Claus, J.J.; van der Cammen, T.J.; Grobbee, D.E.; Hofman, A. Prevalence of Alzheimer’s disease and vascular dementia: Association with education. The Rotterdam study. BMJ 1995, 310, 970–973. [Google Scholar] [CrossRef] [Green Version]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk. Manag. 2019, 15, 259–281. [Google Scholar] [CrossRef]
- Guo, S.; Deng, W.; Xing, C.; Zhou, Y.; Ning, M.; Lo, E.H. Effects of aging, hypertension and diabetes on the mouse brain and heart vasculomes. Neurobiol. Dis. 2019, 126, 117–123. [Google Scholar] [CrossRef]
- Knopman, D.S.; Parisi, J.E.; Boeve, B.F.; Cha, R.H.; Apaydin, H.; Salviati, A.; Edland, S.D.; Rocca, W.A. Vascular dementia in a population-based autopsy study. Arch. Neurol. 2003, 60, 569–575. [Google Scholar] [CrossRef]
- McAleese, K.E.; Alafuzoff, I.; Charidimou, A.; De Reuck, J.; Grinberg, L.T.; Hainsworth, A.H.; Hortobagyi, T.; Ince, P.; Jellinger, K.; Gao, J.; et al. Post-mortem assessment in vascular dementia: Advances and aspirations. BMC Med. 2016, 14, 129. [Google Scholar] [CrossRef]
- Bryant, A.G.; Hu, M.; Carlyle, B.C.; Arnold, S.E.; Frosch, M.P.; Das, S.; Hyman, B.T.; Bennett, R.E. Cerebrovascular Senescence Is Associated With Tau Pathology in Alzheimer’s Disease. Front. Neurol. 2020, 11, 575953. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amyloid Seed | Congo Red Staining | Electron Microscopy | Time to Form Amyloid Fibrils |
|---|---|---|---|
| Synthetic peptide | |||
| Medin-aa 1–50 | + | + | 5–7 days |
| Medin-aa 11–50 | + | + | 8–10 days |
| Medin-aa 31–50 | + | + | 1 day |
| Medin-aa 21–41 | + | + | Instantly |
| Medin-aa 42–49 | + | + | 2–4 days |
| Medin-aa 42–50 | + | + | Instantly |
| Recombinant peptide Medin-aa 1–50 | + | + | Instantly |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; McGraw, K.R.; Monticone, R.E. Milk Fat Globule Epidermal Growth Factor VIII Fragment Medin in Age-Associated Arterial Adverse Remodeling and Arterial Disease. Cells 2023, 12, 253. https://doi.org/10.3390/cells12020253
Wang M, McGraw KR, Monticone RE. Milk Fat Globule Epidermal Growth Factor VIII Fragment Medin in Age-Associated Arterial Adverse Remodeling and Arterial Disease. Cells. 2023; 12(2):253. https://doi.org/10.3390/cells12020253
Chicago/Turabian StyleWang, Mingyi, Kimberly R. McGraw, and Robert E. Monticone. 2023. "Milk Fat Globule Epidermal Growth Factor VIII Fragment Medin in Age-Associated Arterial Adverse Remodeling and Arterial Disease" Cells 12, no. 2: 253. https://doi.org/10.3390/cells12020253