

Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy

{kind=link}
{kind=link}
Abstract
:1. Introduction
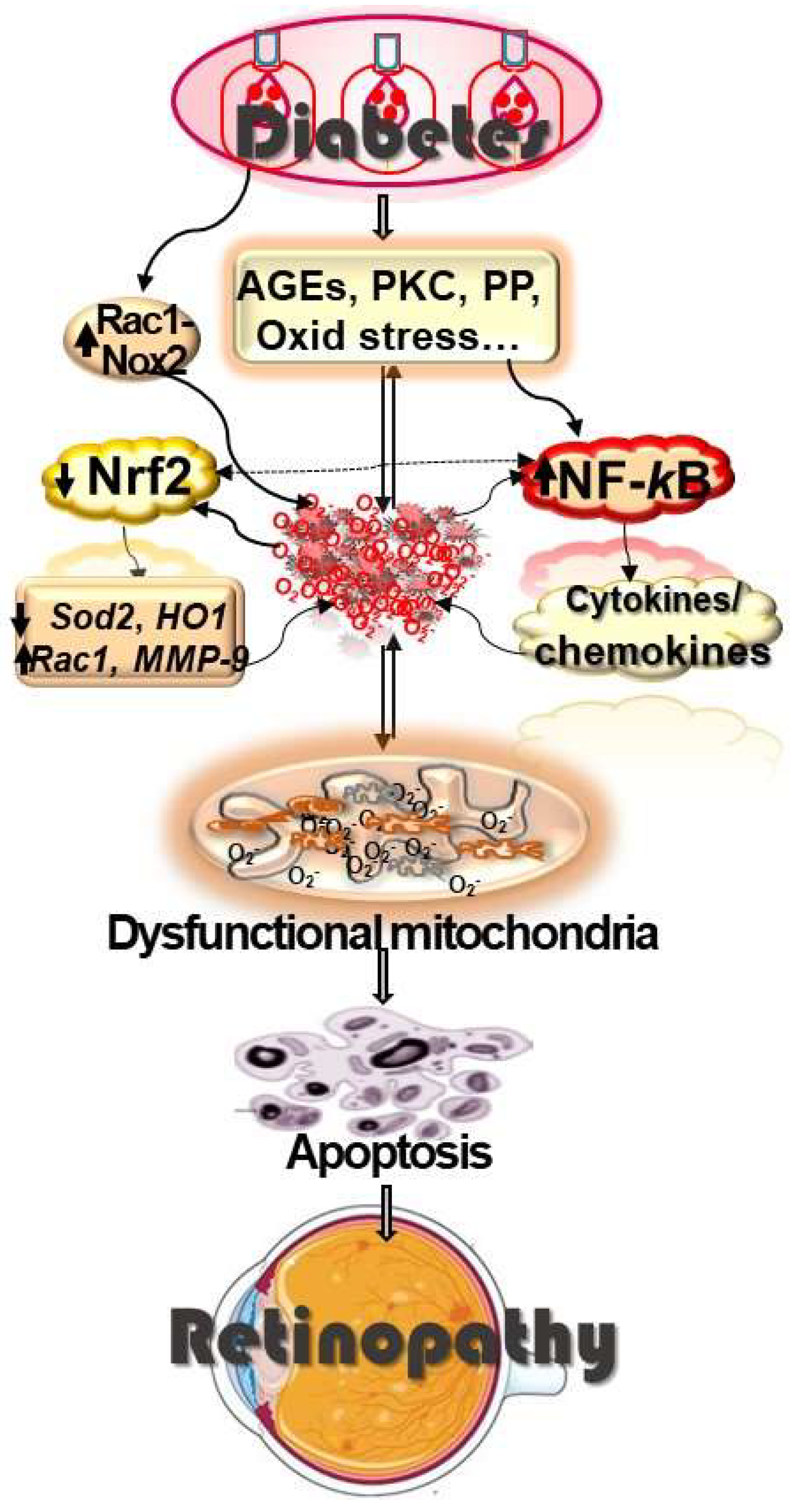
2. Diabetic Retinopathy
2.1. Oxidative Stress and Diabetic Retinopathy
2.2. Inflammation and Diabetic Retinopathy
2.3. Oxidative Stress and Inflammation
3. Epigenetics
3.1. DNA Methylation
3.2. Histone Modifications
3.3. Noncoding RNAs
4. Epigenetics, Oxidative Stress and Diabetic Retinopathy
5. Epigenetics, Inflammation and Diabetic Retinopathy
6. Cross-Talks between Epigenetics, Oxidative Stress and Inflammation
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bommer, C.; Sagalova, V.; Heesemann, E.; Manne-Goehler, J.; Atun, R.; Bärnighausen, T.; Davies, J.; Vollmer, S. Global Economic Burden of Diabetes in Adults: Projections From 2015 to 2030. Diabetes Care 2018, 41, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergul, A.; Kelly-Cobbs, A.; Abdalla, M.; Fagan, S.C. Cerebrovascular complications of diabetes: Focus on stroke. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, M.D.; Brunson, A.; Hedayati, N.; Romano, P.; Melnkow, J. Amputation Risk in Patients with Diabetes Mellitus and Peripheral Artery Disease Using Statewide Data. Ann. Vasc. Surg. 2016, 30, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic retinopathy and systemic factors. Middle East Afr. J. Ophthalmol. 2015, 22, 151–156. [Google Scholar] [CrossRef]
- Maric-Bilkan, C. Sex differences in micro- and macro-vascular complications of diabetes mellitus. Clin. Sci. 2017, 131, 833–846. [Google Scholar] [CrossRef]
- Logette, E.; Lorin, C.; Favreau, C.; Oshurko, E.; Coggan, J.S.; Casalegno, F.; Sy, M.F.; Monney, C.; Bertschy, M.; Delattre, E.; et al. A Machine-Generated View of the Role of Blood Glucose Levels in the Severity of COVID-19. Front. Public Health 2021, 9, 695139. [Google Scholar] [CrossRef]
- Viswanath, K.; McGavin, D.D. Diabetic retinopathy: Clinical findings and management. Community Eye Health 2003, 16, 21–24. [Google Scholar]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A. The neuroscience of diabetic retinopathy. Vis. Neurosci. 2021, 38, E001. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy: Mitochondrial dysfunction and retinal capillary cell death. Antioxid. Redox Signal. 2005, 7, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G. Epigenetic modifications in diabetes. Metabolism 2021, 126, 154920. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Odenbach, S. Role of interleukin-1beta in the development of retinopathy in rats: Effect of antioxidants. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4161–4166. [Google Scholar] [CrossRef] [Green Version]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radic. Biol. Med. 1997, 22, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Zhao, T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid. Redox Signal. 2006, 8, 1533–1548. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 2011, 81, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A.; Zhong, Q.; Santos, J.M. Matrix metalloproteinases in diabetic retinopathy: Potential role of MMP-9. Expert Opin. Investig. Drugs 2012, 21, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A.; Mishra, M. Regulation of matrix metalloproteinase in the pathogenesis of diabetic retinopathy. Prog. Mol. Biol. Transl. Sci. 2017, 148, 67–85. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Tewari, S.; Kowluru, R.A. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic. Biol. Med. 2012, 53, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Diabetic retinopathy, oxidative stress and antioxidants. Curr. Top. Nutraceutical Res. 2005, 3, 209–218. [Google Scholar]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Koppolu, P.; Chakrabarti, S.; Chen, S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic. Res. 2003, 37, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- El-Asrar, A.M. Role of inflammation in the pathogenesis of diabetic retinopathy. Middle East Afr. J. Ophthalmol. 2012, 19, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [Green Version]
- Yuuki, T.; Kanda, T.; Kimura, Y.; Kotajima, N.; Tamura, J.; Kobayashi, I.; Kishi, S. Inflammatory cytokines in vitreous fluid and serum of patients with diabetic vitreoretinopathy. J. Diabetes Its Complicat. 2001, 15, 257–259. [Google Scholar] [CrossRef]
- Meleth, A.D.; Agrón, E.; Chan, C.C.; Reed, G.F.; Arora, K.; Byrnes, G.; Csaky, K.G.; Ferris, F.L., 3rd; Chew, E.Y. Serum inflammatory markers in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4295–4301. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.C.; Allen, D.M.; Cardona, A.E. The Role of Microglia in Diabetic Retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef] [Green Version]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a key event in cancer development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Hilliard, A.; Mendonca, P.; Russell, T.D.; Soliman, K.F.A. The Protective Effects of Flavonoids in Cataract Formation through the Activation of Nrf2 and the Inhibition of MMP-9. Nutrients 2020, 12, 3651. [Google Scholar] [CrossRef]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: They’re not just for matrix anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef]
- Radosinska, J.; Barancik, M.; Vrbjar, N. Heart failure and role of circulating MMP-2 and MMP-9. Panminerva Med. 2017, 59, 241–253. [Google Scholar] [CrossRef]
- Lee, C.Z.; Xue, Z.; Zhu, Y.; Yang, G.Y.; Young, W.L. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke 2007, 38, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-kB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Mishra, M.; Zhong, Q.; Kowluru, R.A. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: Implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic. Biol. Med. 2014, 75, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wei, H.; Song, Y.; Chen, M.; Fan, Z.; Qiu, R.; Zhu, W.; Xu, W.; Wang, F. NF-κB and Keap1 Interaction Represses Nrf2-Mediated Antioxidant Response in Rabbit Hemorrhagic Disease Virus Infection. J. Virol. 2020, 94, e00016-20. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]
- Abcouwer, S.F. Angiogenic Factors and Cytokines in Diabetic Retinopathy. J. Clin. Cell. Immunol. 2013, 11 (Suppl. 1), 1–12. [Google Scholar] [CrossRef] [Green Version]
- Szabó, K.; Énzsöly, A.; Dékány, B.; Szabó, A.; Hajdú, R.I.; Radovits, T.; Mátyás, C.; Oláh, A.; Laurik, L.K.; Somfai, G.M.; et al. Histological Evaluation of Diabetic Neurodegeneration in the Retina of Zucker Diabetic Fatty (ZDF) Rats. Sci. Rep. 2017, 7, 8891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.L.; Li, Z.; Yuan, M.; Yu, A.C.; Zhu, X.; Tso, M.O. Sinomenine inhibits activation of rat retinal microglia induced by advanced glycation end products. Int. Immunopharmacol. 2007, 7, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrósio, A.F.; Fernandes, R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Szyf, M.; Meaney, M.J. Epigenetics, behaviour, and health. Allergy Asthma Clin. Immunol. 2008, 4, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Norris, K.F.; Wang, R.Y.; Kuo, K.C.; Gehrke, C.W. DNA cytosine methylation and heat-induced deamination. Biosci. Rep. 1986, 6, 387–393. [Google Scholar] [CrossRef]
- Jiang, C.; Han, L.; Su, B.; Li, W.H.; Zhao, Z. Features and trend of loss of promoter-associated CpG islands in the human and mouse genomes. Mol. Biol. Evol. 2007, 24, 1991–2000. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Jiang, C. Methylation-dependent transition rates are dependent on local sequence lengths and genomic regions. Mol. Biol. Evol. 2007, 24, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Xu, Y.M.; Du, J.Y.; Lau, A.T. Posttranslational modifications of human histone H3: An update. Proteomics 2014, 14, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell. 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Bär, C.; Chatterjee, S.; Thum, T. Long Noncoding RNAs in Cardiovascular Pathology, Diagnosis, and Therapy. Circulation 2016, 134, 1484–1499. [Google Scholar] [CrossRef]
- Duthie, S.J. Epigenetic modifications and human pathologies: Cancer and CVD. Proc. Nutr. Soc. 2011, 70, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, A.; Riazi-Rad, F.; Havaskary, M.; Nuri, F. Long noncoding RNAs: Regulation, function and cancer. Biotechnol. Genet. Eng. Rev. 2018, 34, 153–180. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, K.M.T.; Elliott, E.K.; Haupt, L.M.; Griffiths, L.R. Regulatory Mechanisms of Epigenetic miRNA Relationships in Human Cancer and Potential as Therapeutic Targets. Cancers 2020, 12, 2922. [Google Scholar] [CrossRef]
- Bure, I.V.; Nemtsova, M.V. Methylation and Noncoding RNAs in Gastric Cancer: Everything Is Connected. Int. J. Mol. Sci. 2021, 22, 5683. [Google Scholar] [CrossRef]
- Maghbooli, Z.; Hossein-Nezhad, A.; Larijani, B.; Amini, M.; Keshtkar, A. Global DNA methylation as a possible biomarker for diabetic retinopathy. Diabetes/Metab. Res. Rev. 2015, 31, 183–189. [Google Scholar] [CrossRef]
- Maghbooli, Z.; Larijani, B.; Emamgholipour, S.; Amini, M.; Keshtkar, A.; Pasalar, P. Aberrant DNA methylation patterns in diabetic nephropathy. J. Diabetes Metab. Disord. 2014, 13, 69. [Google Scholar] [CrossRef] [Green Version]
- Agardh, E.; Lundstig, A.; Perfilyev, A.; Volkov, P.; Freiburghaus, T.; Lindholm, E.; Ronn, T.; Agardh, C.D.; Ling, C. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015, 13, 182. [Google Scholar] [CrossRef] [Green Version]
- Duraisamy, A.J.; Radhakrishnan, R.; Seyoum, B.; Abrams, G.W.; Kowluru, R.A. Epigenetic Modifications in Peripheral Blood as Potential Noninvasive Biomarker of Diabetic Retinopathy. Transl. Vis. Sci. Technol. 2019, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Mishra, M. Therapeutic targets for altering mitochondrial dysfunction associated with diabetic retinopathy. Expert Opin. Ther. Targets 2018, 22, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mohammad, G. Epigenetics and Mitochondrial Stability in the Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Sci. Rep. 2020, 10, 6655. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Kowluru, R.A. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 2011, 60, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Kowluru, R.A. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 2013, 62, 2559–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M.; Zhong, Q.; Kowluru, R.A. Epigenetic modifications of Keap1 regulate its interaction with the protective factor Nrf2 in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7256–7265. [Google Scholar] [CrossRef]
- Zhao, X.; Ling, F.; Zhang, G.W.; Yu, N.; Yang, J.; Xin, X.Y. The Correlation Between MicroRNAs and Diabetic Retinopathy. Front. Immunol. 2022, 13, 941982. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, Z.W.; Wang, Y.; Fu, Y.H.; Gao, X.Y. MicroRNAs: Potential Targets in Diabetic Retinopathy. Horm. Metab. Res. 2020, 52, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, R.; Kowluru, R.A. Long noncoding RNA MALAT1 and Regulation of the Antioxidant Defense System in Diabetic Retinopathy. Diabetes 2021, 70, 227–239. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Nuclear genome-encoded long noncoding RNAs and mitochondrial damage in diabetic retinopathy. Cells 2021, 10, 3271. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, X.; Liao, N.; Ji, Y.; Mi, L.; Gan, Y.; Su, Y.; Wen, F. Identification of NLRP3 Inflammation-Related Gene Promoter Hypomethylation in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2020, 61, 12. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Ye, E.A.; Liu, L.; Jiang, Y.; Jan, J.; Gaddipati, S.; Suvas, S.; Steinle, J.J. miR-15a/16 reduces retinal leukostasis through decreased pro-inflammatory signaling. J. Neuroinflammation 2016, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.Y.; Yu, F.; Bayless, K.J.; Ko, M.L. MicroRNA-150 (miR-150) and Diabetic Retinopathy: Is miR-150 Only a Biomarker or Does It Contribute to Disease Progression? Int. J. Mol. Sci. 2022, 23, 12099. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Navitskaya, S.; Chakravarthy, H.; Huang, C.; Kady, N.; Lydic, T.A.; Chen, Y.E.; Yin, K.J.; Powell, F.L.; Martin, P.M.; et al. Dual Anti-Inflammatory and Anti-Angiogenic Action of miR-15a in Diabetic Retinopathy. EBioMedicine 2016, 11, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Jiang, X.; Tong, T.; Chang, H.; Li, R.X. MiR-204 inhibits inflammation and cell apoptosis in retinopathy rats with diabetic retinopathy by regulating Bcl-2 and SIRT1 expressions. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Thomas, A.A.; Chen, S.; Aref-Eshghi, E.; Feng, B.; Gonder, J.; Sadikovic, B.; Chakrabarti, S. MALAT1: An Epigenetic Regulator of Inflammation in Diabetic Retinopathy. Sci. Rep. 2018, 8, 6526. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.A.; Feng, B.; Chakrabarti, S. ANRIL: A Regulator of VEGF in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 470–480. [Google Scholar] [CrossRef]
- Biswas, S.; Feng, B.; Chen, S.; Liu, J.; Aref-Eshghi, E.; Gonder, J.; Ngo, V.; Sadikovic, B.; Chakrabarti, S. The Long Non-Coding RNA HOTAIR Is a Critical Epigenetic Mediator of Angiogenesis in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 20. [Google Scholar] [CrossRef]
- Geraldes, P.; Hiraoka-Yamamoto, J.; Matsumoto, M.; Clermont, A.; Leitges, M.; Marette, A.; Aiello, L.P.; Kern, T.S.; King, G.L. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 2009, 15, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Shan, Y.; Mishra, M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab. Investig. 2016, 96, 1040–1049. [Google Scholar] [CrossRef] [Green Version]
- Duraisamy, A.J.; Mishra, M.; Kowluru, A.; Kowluru, R.A. Epigenetics and regulation of oxidative stress in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4831–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M.; Kowluru, R.A. Role of PARP-1 as a novel transcriptional regulator of MMP-9 in diabetic retinopathy. Biochim. Et Biophys. Acta 2017, 1863, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, J.; Chen, X.; Yang, Y.; Wang, F.; Li, W.; Awuti, M.; Sun, Y.; Lian, C.; Li, Z.; et al. miR-365 promotes diabetic retinopathy through inhibiting Timp3 and increasing oxidative stress. Exp. Eye Res. 2018, 168, 89–99. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Park, J.; Dang, T.L.; Choi, Y.G.; Kim, V.N. Microprocessor depends on hemin to recognize the apical loop of primary microRNA. Nucleic Acids Res. 2018, 46, 5726–5736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, P.; Muraleedharan, C.K.; Xu, S. Intraocular Delivery of miR-146 Inhibits Diabetes-Induced Retinal Functional Defects in Diabetic Rat Model. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolling, M.; Genschel, C.; Kaucsar, T.; Hubner, A.; Rong, S.; Schmitt, R.; Sorensen-Zender, I.; Haddad, G.; Kistler, A.; Seeger, H.; et al. Hypoxia-induced long non-coding RNA Malat1 is dispensable for renal ischemia/reperfusion-injury. Sci. Rep. 2018, 8, 3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.; Xu, B.; Shi, H. Long noncoding RNA MALAT1 acts as a competing endogenous RNA to regulate Amadori-glycated albumin-induced MCP-1 expression in retinal microglia by a microRNA-124-dependent mechanism. Inflamm. Res. 2018, 67, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Hu, H.Y.; You, Z.P.; Li, B.Y.; Shi, K. Targeting long non-coding RNA MALAT1 alleviates retinal neurodegeneration in diabetic mice. Int. J. Ophthalmol. 2020, 13, 213–219. [Google Scholar] [CrossRef]
- Thomas, A.A.; Biswas, S.; Feng, B.; Chen, S.; Gonder, J.; Chakrabarti, S. lncRNA H19 prevents endothelial-mesenchymal transition in diabetic retinopathy. Diabetologia 2019, 62, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, M.; Chen, J.; Lin, S.; Cai, D.; Chen, C.; Chen, Z. Long non-coding RNA MIAT acts as a biomarker in diabetic retinopathy by absorbing miR-29b and regulating cell apoptosis. Biosci. Rep. 2017, 37, BSR20170036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Ye, Q.; Ning, S.; Yang, Z.; Chen, Y.; Zhang, L.; Huang, Y.; Xie, F.; Cheng, X.; Chi, J.; et al. LncRNA MEG3 promotes endoplasmic reticulum stress and suppresses proliferation and invasion of colorectal carcinoma cells through the MEG3/miR-103a-3p/PDHB ceRNA pathway. Neoplasma 2021, 68, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qin, H.; Leng, Y.; Li, X.; Zhang, L.; Bai, D.; Meng, Y.; Wang, J. LncRNA MEG3 overexpression inhibits the development of diabetic retinopathy by regulating TGF-β1 and VEGF. Exp. Ther. Med. 2018, 16, 2337–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressler, S.B.; Liu, D.; Glassman, A.R.; Blodi, B.A.; Castellarin, A.A.; Jampol, L.M.; Kaufman, P.L.; Melia, M.; Singh, H.; Wells, J.A. Change in Diabetic Retinopathy Through 2 Years: Secondary Analysis of a Randomized Clinical Trial Comparing Aflibercept, Bevacizumab, and Ranibizumab. JAMA Ophthalmol. 2017, 135, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.V.; Husain, D. Panretinal Photocoagulation: A Review of Complications. Semin. Ophthalmol. 2018, 33, 83–88. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowluru, R.A. Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy. Cells 2023, 12, 300. https://doi.org/10.3390/cells12020300
Kowluru RA. Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy. Cells. 2023; 12(2):300. https://doi.org/10.3390/cells12020300
Chicago/Turabian StyleKowluru, Renu A. 2023. "Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy" Cells 12, no. 2: 300. https://doi.org/10.3390/cells12020300
APA StyleKowluru, R. A. (2023). Cross Talks between Oxidative Stress, Inflammation and Epigenetics in Diabetic Retinopathy. Cells, 12(2), 300. https://doi.org/10.3390/cells12020300