
The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored

 ,
,  ,
,  , , , and
, , , and
Abstract
:1. HNF1B Deficiency: Genetics and Historical Background
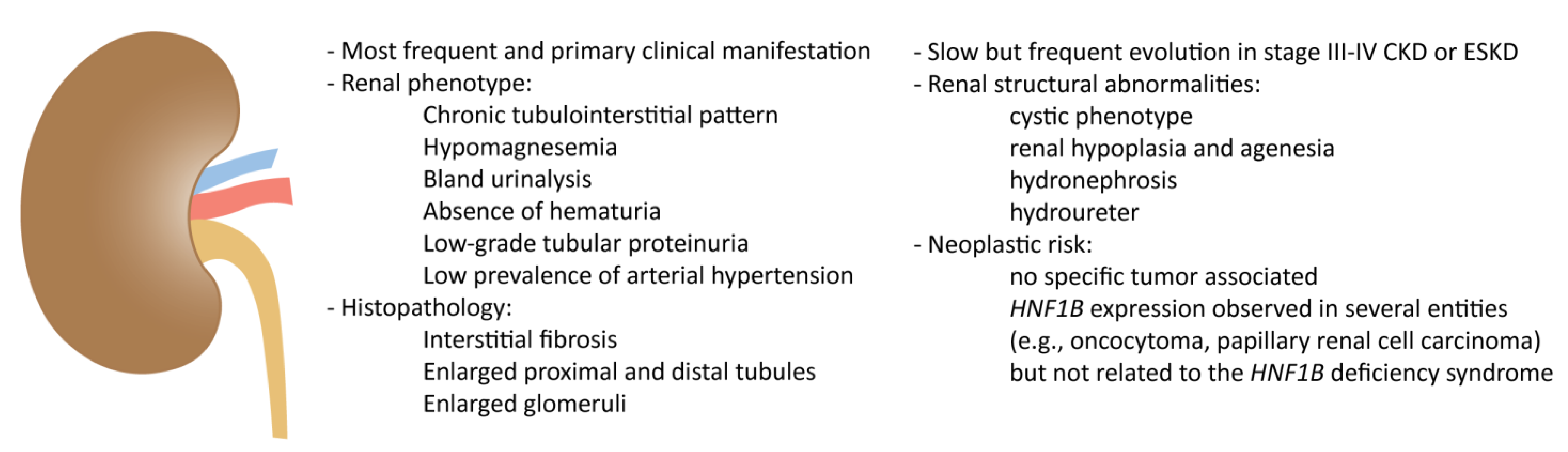
2. Kidney Involvement in HNF1B Deficiency
2.1. Clinical Spectrum
2.2. Histopathology of Non-Neoplastic Conditions
2.3. Histopathology of Neoplastic Conditions
3. Liver Involvement in HNF1B Deficiency
3.1. Clinical Spectrum
3.2. Histopathology of Non-Neoplastic Conditions
3.3. Histopathology of Neoplastic Conditions
4. Pancreas Involvement in HNF1B Deficiency
4.1. Clinical Spectrum
4.2. Histopathology of Non-Neoplastic Condition
4.3. Histopathology of Neoplastic Condition
5. Additional Extrarenal Involvement
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGS | Alagille syndrome |
| BDCs | bile duct cysts |
| BMI | body mass index |
| BW | birth weight |
| CKD | chronic kidney disease |
| DCDC2 | doublecortin domain containing protein 2 |
| DPMs | ductal plate malformations |
| EASL | European Association for the Study of the Liver |
| ESKD | end-stage kidney disease |
| HCC | hepatocellular carcinoma |
| HNF | hepatocyte nuclear factor |
| HNF1B | hepatocyte nuclear factor 1β |
| IBATis | ileal bile acid transporter inhibitors |
| IUGR | intrauterine growth restriction |
| KIF12 | kinesin family member 12 |
| MeSH | Medical Subject Headings |
| NA | information not available |
| NSC | neonatal sclerosing cholangitis |
| PDS | primitive ductal structures |
| PFIC | progressive familial intrahepatic cholestasis |
| PILBD | paucity of intralobular bile ducts |
| RSA | renal structural abnormalities |
| SGA | small for gestational age |
| SNPs | single nucleotide polymorphisms |
| TCF2 | transcription factor 2 |
| US | ultrasound |
References
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The molecular functions of hepatocyte nuclear factors—In and beyond the liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef] [Green Version]
- Bockenhauer, D.; Jaureguiberry, G. HNF1B-associated clinical phenotypes: The kidney and beyond. Pediatr. Nephrol. 2016, 31, 707–714. [Google Scholar] [CrossRef]
- El-Khairi, R.; Vallier, L. The role of hepatocyte nuclear factor 1beta in disease and development. Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 23–32. [Google Scholar] [CrossRef] [Green Version]
- Nyunt, O.; Wu, J.Y.; McGown, I.N.; Harris, M.; Huynh, T.; Leong, G.M.; Cowley, D.M.; Cotterill, A.M. Investigating maturity onset diabetes of the young. Clin. Biochem. Rev. 2009, 30, 67–74. [Google Scholar]
- Verhave, J.C.; Bech, A.P.; Wetzels, J.F.; Nijenhuis, T. Hepatocyte Nuclear Factor 1beta-Associated Kidney Disease: More than Renal Cysts and Diabetes. J. Am. Soc. Nephrol. 2016, 27, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Clissold, R.L.; Hamilton, A.J.; Hattersley, A.T.; Ellard, S.; Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 2015, 11, 102–112. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Pal, A.; Reidy, K.J. Genetic Syndromes Affecting Kidney Development. Results Probl. Cell Differ. 2017, 60, 257–279. [Google Scholar]
- Mitchel, M.W.; Moreno-De-Luca, D.; Myers, S.M.; Levy, R.V.; Turner, S.; Ledbetter, D.H.; Martin, C.L. 17q12 Recurrent Deletion Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK401562/2020 (accessed on 15 October 2020).
- Faguer, S.; Decramer, S.; Chassaing, N.; Bellanné-Chantelot, C.; Calvas, P.; Beaufils, S.; Bessenay, L.; Lengelé, J.P.; Dahan, K.; Ronco, P.; et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011, 80, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Bellanné-Chantelot, C.; Clauin, S.; Chauveau, D.; Collin, P.; Daumont, M.; Douillard, C.; Dubois-Laforgue, D.; Dusselier, L.; Gautier, J.F.; Jadoul, M.; et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 2005, 54, 3126–3132. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Laforgue, D.; Cornu, E.; Saint-Martin, C.; Coste, J.; Bellanné-Chantelot, C.; Timsit, J.; Monogenic Diabetes Study Group of the Société Francophone du Diabète. Diabetes, Associated Clinical Spectrum, Long-term Prognosis, and Genotype/Phenotype Correlations in 201 Adult Patients with Hepatocyte Nuclear Factor 1B (HNF1B) Molecular Defects. Diabetes Care 2017, 40, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, A.; Corveleyn, A.; Devriendt, K.; van Tienoven, T.P.; Allegaert, K.; Van Dyck, M.; van den Heuvel, L.; Kuypers, D.; Claes, K.; Mekahli, D.; et al. Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol. Dial. Transplant. 2015, 30, 835–842. [Google Scholar] [CrossRef]
- Musetti, C.; Quaglia, M.; Mellone, S.; Pagani, A.; Fusco, I.; Monzani, A.; Giordano, M.; Stratta, P. Chronic renal failure of unknown origin is caused by HNF1B mutations in 9% of adult patients: A single centre cohort analysis. Nephrology 2014, 19, 202–209. [Google Scholar] [CrossRef]
- Roelandt, P.; Antoniou, A.; Libbrecht, L.; Van Steenbergen, W.; Laleman, W.; Verslype, C.; Van der Merwe, S.; Nevens, F.; De Vos, R.; Fischer, E.; et al. HNF1B deficiency causes ciliary defects in human cholangiocytes. Hepatology 2012, 56, 1178–1181. [Google Scholar] [CrossRef]
- Montoli, A.; Colussi, G.; Massa, O.; Caccia, R.; Rizzoni, G.; Civati, G.; Barbetti, F. Renal cysts and diabetes syndrome linked to mutations of the hepatocyte nuclear factor-1 beta gene: Description of a new family with associated liver involvement. Am. J. Kidney Dis. 2002, 40, 397–402. [Google Scholar] [CrossRef]
- Sagen, J.V.; Bostad, L.; Njolstad, P.R.; Sovik, O. Enlarged nephrons and severe nondiabetic nephropathy in hepatocyte nuclear factor-1beta (HNF-1beta) mutation carriers. Kidney Int. 2003, 64, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Piedrafita, A.; Balayssac, S.; Casemayou, A.; Saulnier-Blache, J.S.; Lucas, A.; Iacovoni, J.S.; Breuil, B.; Chauveau, D.; Decramer, S.; Malet-Martino, M.; et al. Hepatocyte nuclear factor-1beta shapes the energetic homeostasis of kidney tubule cells. FASEB J. 2021, 35, e21931. [Google Scholar] [CrossRef]
- Adalat, S.; Woolf, A.S.; Johnstone, K.A.; Wirsing, A.; Harries, L.W.; Long, D.A.; Hennekam, R.C.; Ledermann, S.E.; Rees, L.; Van′t Hoff, W.; et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J. Am. Soc. Nephrol. 2009, 20, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Frindt, G.; Palmer, L.G. Magnesium modulates ROMK channel-mediated potassium secretion. J. Am. Soc. Nephrol. 2010, 21, 2109–2116. [Google Scholar] [CrossRef] [Green Version]
- Van Der Made, C.I.; Hoorn, E.J.; De La Faille, R.; Karaaslan, H.; Knoers, N.V.; Hoenderop, J.G.; Poussou, R.V.; De Baaij, J.H. Hypomagnesemia as First Clinical Manifestation of ADTKD-HNF1B: A Case Series and Literature Review. Am. J. Nephrol. 2015, 42, 85–90. [Google Scholar] [CrossRef]
- Musetti, C.; Quaglia, M.; Stratta, P.; Giordano, M. Hypomagnesemia and progressive chronic kidney disease: Thinking of HNF1B and other genetic nephropathies. Kidney Int. 2015, 88, 641. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.C.; Zhang, Y.; Shao, A.; Avdulov, S.; Herrera, J.; Aboudehen, K.; Pontoglio, M.; Igarashi, P. Mechanism of Fibrosis in HNF1B-Related Autosomal Dominant Tubulointerstitial Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2493–2509. [Google Scholar] [CrossRef]
- Ge, S.; Yang, M.; Cui, Y.; Wu, J.; Xu, L.; Dong, J.; Liao, L. The Clinical Characteristics and Gene Mutations of Maturity-Onset Diabetes of the Young Type 5 in Sixty-One Patients. Front. Endocrinol. 2022, 13, 911526. [Google Scholar] [CrossRef]
- Hua Tan, C.S.; Ang, S.F.; Yeoh, E.; Goh, B.X.; Loh, W.J.; Shum, C.F.; May Ping Eng, M.; Yan Lun Liu, A.; Wan Ting Chan, L.; Goh, L.X.; et al. MODY5 Hepatocyte Nuclear Factor 1ss (HNF1ss)-Associated Nephropathy: Experience from a regional monogenic diabetes referral centre in Singapore. J. Investig. Med. High Impact Case Rep. 2022, 10, 23247096211065626. [Google Scholar] [CrossRef]
- Bingham, C.; Ellard, S.; Allen, L.; Bulman, M.; Shepherd, M.; Frayling, T.; Berry, P.J.; Clark, P.M.; Lindner, T.; Bell, G.I.; et al. Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1 beta. Kidney Int. 2000, 57, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Horster, M.F.; Braun, G.S.; Huber, S.M. Embryonic renal epithelia: Induction, nephrogenesis, and cell differentiation. Physiol. Rev. 1999, 79, 1157–1191. [Google Scholar] [CrossRef] [Green Version]
- Lazzaro, D.; De Simone, V.; De Magistris, L.; Lehtonen, E.; Cortese, R. LFB1 and LFB3 homeoproteins are sequentially expressed during kidney development. Development 1992, 114, 469–479. [Google Scholar] [CrossRef]
- Lokmane, L.; Heliot, C.; Garcia-Villalba, P.; Fabre, M.; Cereghini, S. vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development 2010, 137, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Lindström, N.O.; McMahon, J.A.; Guo, J.; Tran, T.; Guo, Q.; Rutledge, E.; Parvez, R.K.; Saribekyan, G.; Schuler, R.E.; Liao, C.; et al. Conserved and Divergent Features of Human and Mouse Kidney Organogenesis. J. Am. Soc. Nephrol. 2018, 29, 785–805. [Google Scholar] [CrossRef] [Green Version]
- Ferre, S.; Igarashi, P. New insights into the role of HNF-1beta in kidney (patho)physiology. Pediatr. Nephrol. 2019, 34, 1325–1335. [Google Scholar] [CrossRef]
- Naylor, R.W.; Davidson, A.J. Hnf1beta and nephron segmentation. Pediatr. Nephrol. 2014, 29, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Heidet, L.; Decramer, S.; Pawtowski, A.; Morinière, V.; Bandin, F.; Knebelmann, B.; Lebre, A.S.; Faguer, S.; Guigonis, V.; Antignac, C.; et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1079–1090. [Google Scholar] [CrossRef]
- Bellanné-Chantelot, C.; Chauveau, D.; Gautier, J.F.; Dubois-Laforgue, D.; Clauin, S.; Beaufils, S.; Wilhelm, J.M.; Boitard, C.; Noël, L.H.; Velho, G.; et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann. Intern. Med. 2004, 140, 510–517. [Google Scholar] [CrossRef]
- Edghill, E.L.; Bingham, C.; Ellard, S.; Hattersley, A.T. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J. Med. Genet. 2006, 43, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Bingham, C.; Bulman, M.P.; Ellard, S.; Allen, L.I.; Lipkin, G.W.; van′t Hoff, W.G.; Woolf, A.S.; Rizzoni, G.; Novelli, G.; Nicholls, A.J.; et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am. J. Hum. Genet. 2001, 68, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Rizzoni, G.; Loirat, C.; Levy, M.; Milanesi, C.; Zachello, G.; Mathieu, H. Familial hypoplastic glomerulocystic kidney. A new entity? Clin. Nephrol. 1982, 18, 263–268. [Google Scholar]
- Kaplan, B.S.; Gordon, I.; Pincott, J.; Barratt, T.M. Familial hypoplastic glomerulocystic kidney disease: A definite entity with dominant inheritance. Am. J. Med. Genet. 1989, 34, 569–573. [Google Scholar] [CrossRef]
- Mache, C.J.; Preisegger, K.H.; Kopp, S.; Ratschek, M.; Ring, E. De novo HNF-1 beta gene mutation in familial hypoplastic glomerulocystic kidney disease. Pediatr. Nephrol. 2002, 17, 1021–1026. [Google Scholar] [CrossRef]
- Hojny, J.; Michalkova, R.; Krkavcova, E.; Bui, Q.H.; Bartu, M.; Nemejcova, K.; Kalousova, M.; Kleiblova, P.; Dundr, P.; Struzinska, I. Comprehensive quantitative analysis of alternative splicing variants reveals the HNF1B mRNA splicing pattern in various tumour and non-tumour tissues. Sci. Rep. 2022, 12, 199. [Google Scholar] [CrossRef]
- Suzuki, E.; Kajita, S.; Takahashi, H.; Matsumoto, T.; Tsuruta, T.; Saegusa, M. Transcriptional upregulation of HNF-1beta by NF-kappaB in ovarian clear cell carcinoma modulates susceptibility to apoptosis through alteration in bcl-2 expression. Lab. Invest. 2015, 95, 962–972. [Google Scholar] [CrossRef]
- Tsuchiya, A.; Sakamoto, M.; Yasuda, J.; Chuma, M.; Ohta, T.; Ohki, M.; Yasugi, T.; Taketani, Y.; Hirohashi, S. Expression profiling in ovarian clear cell carcinoma: Identification of hepatocyte nuclear factor-1 beta as a molecular marker and a possible molecular target for therapy of ovarian clear cell carcinoma. Am. J. Pathol. 2003, 163, 2503–2512. [Google Scholar] [CrossRef]
- Buchner, A.; Castro, M.; Hennig, A.; Popp, T.; Assmann, G.; Stief, C.G.; Zimmermann, W. Downregulation of HNF-1B in renal cell carcinoma is associated with tumor progression and poor prognosis. Urology 2010, 76, 507 e6-11. [Google Scholar] [CrossRef]
- Köbel, M.; Kalloger, S.E.; Carrick, J.; Huntsman, D.; Asad, H.; Oliva, E.; Ewanowich, C.A.; Soslow, R.A.; Gilks, C.B. A limited panel of immunomarkers can reliably distinguish between clear cell and high-grade serous carcinoma of the ovary. Am. J. Surg. Pathol. 2009, 33, 14–21. [Google Scholar] [CrossRef]
- Amano, Y.; Mandai, M.; Yamaguchi, K.; Matsumura, N.; Kharma, B.; Baba, T.; Abiko, K.; Hamanishi, J.; Yoshioka, Y.; Konishi, I. Metabolic alterations caused by HNF1beta expression in ovarian clear cell carcinoma contribute to cell survival. Oncotarget 2015, 6, 26002–26017. [Google Scholar] [CrossRef] [Green Version]
- Cuff, J.; Salari, K.; Clarke, N.; Esheba, G.E.; Forster, A.D.; Huang, S.; West, R.B.; Higgins, J.P.; Longacre, T.A.; Pollack, J.R. Integrative bioinformatics links HNF1B with clear cell carcinoma and tumor-associated thrombosis. PLoS ONE 2013, 8, e74562. [Google Scholar] [CrossRef] [Green Version]
- Němejcová, K.; Tichá, I.; Kleiblová, P.; Bártů, M.; Cibula, D.; Jirsová, K.; Dundr, P. Expression, Epigenetic and Genetic Changes of HNF1B in Endometrial Lesions. Pathol. Oncol. Res. 2016, 22, 523–530. [Google Scholar] [CrossRef]
- Lebrun, G.; Vasiliu, V.; Bellanné-Chantelot, C.; Bensman, A.; Ulinski, T.; Chrétien, Y.; Grünfeld, J.P. Cystic kidney disease, chromophobe renal cell carcinoma and TCF2 (HNF1 beta) mutations. Nat. Clin. Pract. Nephrol. 2005, 1, 115–119. [Google Scholar] [CrossRef]
- Bártů, M.; Hojný, J.; Hájková, N.; Michálková, R.; Krkavcová, E.; Hadravský, L.; Kleissnerová, L.; Bui, Q.H.; Stružinská, I.; Němejcová, K.; et al. Analysis of expression, epigenetic, and genetic changes of HNF1B in 130 kidney tumours. Sci. Rep. 2020, 10, 17151. [Google Scholar] [CrossRef]
- Chandra, S.; Srinivasan, S.; Batra, J. Hepatocyte nuclear factor 1 beta: A perspective in cancer. Cancer Med. 2021, 10, 1791–1804. [Google Scholar] [CrossRef]
- An, J.; Park, C.K.; Kim, M.; Joo, J.W.; Cho, N.H. HNF-1beta as an immunohistochemical marker for distinguishing chromophobe renal cell carcinoma and hybrid oncocytic tumors from renal oncocytoma. Virchows Arch. 2021, 478, 459–470. [Google Scholar] [CrossRef]
- Kato, N.; Motoyama, T. Hepatocyte nuclear factor-1beta(HNF-1beta) in human urogenital organs: Its expression and role in embryogenesis and tumorigenesis. Histol. Histopathol. 2009, 24, 1479–1486. [Google Scholar]
- Wang, C.C.; Mao, T.L.; Yang, W.C.; Jeng, Y.M. Underexpression of hepatocyte nuclear factor-1beta in chromophobe renal cell carcinoma. Histopathology 2013, 62, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.R.; Hirsch, M.S.; Jo, V.Y. HNF1beta and S100A1 are useful biomarkers for distinguishing renal oncocytoma and chromophobe renal cell carcinoma in FNA and core needle biopsies. Cancer Cytopathol. 2015, 123, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Tong, P.; Kong, W.; Dong, B.; Huang, Y.; Park, I.Y.; Zhou, L.; Liu, X.D.; Ding, Z.; Zhang, X.; et al. HNF1B Loss Exacerbates the Development of Chromophobe Renal Cell Carcinomas. Cancer Res. 2017, 77, 5313–5326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kanyomse, Q.; Xie, Y. Tumor-suppressive activity of Hnf1beta in Wilms’ tumor. Biosci. Biotechnol. Biochem. 2019, 83, 2008–2015. [Google Scholar] [CrossRef]
- Bártů, M.; Dundr, P.; Němejcová, K.; Tichá, I.; Hojný, H.; Hájková, N. The Role of HNF1B in Tumorigenesis of Solid Tumours: A Review of Current Knowledge. Folia Biol. 2018, 64, 71–83. [Google Scholar]
- Szponar, A.; Yusenko, M.V.; Kuiper, R.; van Kessel, A.G.; Kovacs, G. Genomic profiling of papillary renal cell tumours identifies small regions of DNA alterations: A possible role of HNF1B in tumour development. Histopathology 2011, 58, 934–943. [Google Scholar] [CrossRef]
- Banyai, D.; Sarlos, D.P.; Nagy, A.; Kovacs, G. Recalling Cohnheim’s Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. Int. J. Biol. Sci. 2018, 14, 784–790. [Google Scholar] [CrossRef]
- Kotalova, R.; Dusatkova, P.; Cinek, O.; Dusatkova, L.; Dedic, T.; Seeman, T.; Lebl, J.; Pruhova, S. Hepatic phenotypes of HNF1B gene mutations: A case of neonatal cholestasis requiring portoenterostomy and literature review. World J. Gastroenterol. 2015, 21, 2550–2557. [Google Scholar] [CrossRef]
- Raile, K.; Klopocki, E.; Holder, M.; Wessel, T.; Galler, A.; Deiss, D.; Muller, D.; Riebel, T.; Horn, D.; Maringa, M.; et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J. Clin. Endocrinol. Metab. 2009, 94, 2658–2664. [Google Scholar] [CrossRef] [Green Version]
- Beckers, D.; Bellanne-Chantelot, C.; Maes, M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta). J. Pediatr. 2007, 150, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Kitanaka, S.; Miki, Y.; Hayashi, Y.; Igarashi, T. Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with Type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J. Clin. Endocrinol. Metab. 2004, 89, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Leusse, C.; De Paula, A.M.; Aschero, A.; Parache, C.; Hery, G.; Cailliez, M.; Missirian, C.; Fabre, A. Hepatocarcinoma and Cholestasis Associated to Germline Hemizygous Deletion of Gene HNF1B. J. Pediatr. Gastroenterol. Nutr. 2019, 68, e85. [Google Scholar] [CrossRef] [PubMed]
- Pinon, M.; Carboni, M.; Colavito, D.; Cisarò, F.; Peruzzi, L.; Pizzol, A.; Calosso, G.; David, E.; Calvo, P.L. Not only Alagille syndrome. Syndromic paucity of interlobular bile ducts secondary to HNF1beta deficiency: A case report and literature review. Ital. J. Pediatr. 2019, 45, 27. [Google Scholar] [CrossRef] [PubMed]
- Mandato, C.; Zollo, G.; Vajro, P. Cholestatic jaundice in infancy: Struggling with many old and new phenotypes. Ital. J. Pediatr. 2019, 45, 83. [Google Scholar] [CrossRef]
- Ünlüsoy Aksu, A.; Das, S.K.; Nelson-Williams, C.; Jain, D.; Özbay Hoşnut, F.; Evirgen Şahin, G.; Lifton, R.P.; Vilarinho, S. Recessive Mutations in KIF12 Cause High Gamma-Glutamyltransferase Cholestasis. Hepatol. Commun. 2019, 3, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 2019, 21, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Stalke, A.; Sgodda, M.; Cantz, T.; Skawran, B.; Lainka, E.; Hartleben, B.; Baumann, U.; Pfister, E.D. KIF12 Variants and Disturbed Hepatocyte Polarity in Children with a Phenotypic Spectrum of Cholestatic Liver Disease. J. Pediatr. 2022, 240, 284–291 e9. [Google Scholar] [CrossRef]
- Gong, Y.; Ma, Z.; Patel, V.; Fischer, E.; Hiesberger, T.; Pontoglio, M.; Igarashi, P. HNF-1beta regulates transcription of the PKD modifier gene Kif12. J. Am. Soc. Nephrol. 2009, 20, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Mrug, M.; Li, R.; Cui, X.; Schoeb, T.R.; Churchill, G.A.; Guay-Woodford, L.M. Kinesin family member 12 is a candidate polycystic kidney disease modifier in the cpk mouse. J. Am. Soc. Nephrol. 2005, 16, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Kamath, B.M.; Stein, P.; Houwen, R.H.J.; Verkade, H.J. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020, 40, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Karpen, S.J.; Kelly, D.; Mack, C.; Stein, P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol. Int. 2020, 14, 677–689. [Google Scholar] [CrossRef]
- Gonzales, E.; Hardikar, W.; Stormon, M.; Baker, A.; Hierro, L.; Gliwicz, D.; Lacaille, F.; Lachaux, A.; Sturm, E.; Setchell, K.D.; et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomised phase 2 study. Lancet 2021, 398, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511. [Google Scholar] [CrossRef]
- Coffinier, C.; Gresh, L.; Fiette, L.; Tronche, F.; Schutz, G.; Babinet, C.; Pontoglio, M.; Yaniv, M.; Barra, J. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development 2002, 129, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Sada, A.; Iwata, T.; Niwa, T.; Tomizawa, M.; Xanthopoulos, K.G.; Koike, T.; Shiojiri, N. Suppression of C/EBPalpha expression in periportal hepatoblasts may stimulate biliary cell differentiation through increased Hnf6 and Hnf1b expression. Development 2006, 133, 4233–4243. [Google Scholar] [CrossRef] [Green Version]
- Clotman, F.; Libbrecht, L.; Gresh, L.; Yaniv, M.; Roskams, T.; Rousseau, G.G.; Lemaigre, F.P. Hepatic artery malformations associated with a primary defect in intrahepatic bile duct development. J. Hepatol. 2003, 39, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, P.; Tate, J.; Callens, C.; Cordi, S.; Vandersmissen, P.; Carpentier, R.; Sempoux, C.; Devuyst, O.; Pierreux, C.E.; Courtoy, P.; et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis. Hepatology 2011, 53, 1959–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimizu, N.; Miyajima, A.; Mostov, K.E. Liver progenitor cells fold up a cell monolayer into a double-layered structure during tubular morphogenesis. Mol. Biol. Cell 2009, 20, 2486–2494. [Google Scholar] [CrossRef] [Green Version]
- Tanimizu, N.; Miyajima, A.; Mostov, K.E. Liver progenitor cells develop cholangiocyte-type epithelial polarity in three-dimensional culture. Mol. Biol. Cell 2007, 18, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, P.; Carpentier, R.; Antoniou, A.; Lemaigre, F.P. Biliary differentiation and bile duct morphogenesis in development and disease. Int. J. Biochem. Cell Biol. 2011, 43, 245–256. [Google Scholar] [CrossRef]
- Gunay-Aygun, M. Liver and kidney disease in ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettunen, J.L.; Parviainen, H.; Miettinen, P.J.; Färkkilä, M.; Tamminen, M.; Salonen, P.; Lantto, E.; Tuomi, T. Biliary Anomalies in Patients With HNF1B Diabetes. J. Clin. Endocrinol. Metab. 2017, 102, 2075–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonc, E.N.; Ozturk, B.B.; Haldorsen, I.S.; Molnes, J.; Immervoll, H.; Ræder, H.; Molven, A.; Søvik, O.; Njølstad, P.R. HNF1B mutation in a Turkish child with renal and exocrine pancreas insufficiency, diabetes and liver disease. Pediatr. Diabetes 2012, 13, e1–e5. [Google Scholar] [CrossRef]
- Francis, Y.; Tiercelin, C.; Alexandre-Heyman, L.; Larger, E.; Dubois-Laforgue, D. HNF1B-MODY Masquerading as Type 1 Diabetes: A Pitfall in the Etiological Diagnosis of Diabetes. J. Endocr. Soc. 2022, 6, bvac087. [Google Scholar] [CrossRef]
- Ayoub, M.D.; Kamath, B.M. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics 2020, 10, 907. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Kamath, B.M.; Loomes, K.M.; Karpen, S.J. Cholestatic liver diseases of genetic etiology: Advances and controversies. Hepatology 2022, 75, 1627–1646. [Google Scholar] [CrossRef]
- Li, J.Q.; Lu, Y.; Qiu, Y.L.; Wang, J.S. Neonatal sclerosing cholangitis caused by DCDC2 variations in two siblings and literature review. Zhonghua Er Ke Za Zhi 2018, 56, 623–627. [Google Scholar]
- Grammatikopoulos, T.; Sambrotta, M.; Strautnieks, S.; Foskett, P.; Knisely, A.S.; Wagner, B.; Deheragoda, M.; Starling, C.; Mieli-Vergani, G.; Smith, J.; et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J. Hepatol. 2016, 65, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Girard, M.; Bizet, A.A.; Lachaux, A.; Gonzales, E.; Filhol, E.; Collardeau-Frachon, S.; Jeanpierre, C.; Henry, C.; Fabre, M.; Viremouneix, L.; et al. DCDC2 Mutations Cause Neonatal Sclerosing Cholangitis. Hum. Mutat. 2016, 37, 1025–1029. [Google Scholar] [CrossRef]
- Khanna, R.; Verma, S.K. Pediatric hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 3980–3999. [Google Scholar] [CrossRef] [PubMed]
- Atra, A.; Al-Asiri, R.; Wali, S.; Al-Husseini, H.; Al-Bassas, A.; Zimmermann, A. Hepatocellular carcinoma, syncytial giant cell: A novel variant in children: A case report. Ann. Diagn. Pathol. 2007, 11, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Pinon, M.; Gambella, A.; Giugliano, L.; Chiadò, C.; Kalantari, S.; Bracciamà, V.; Deaglio, S.; Tinti, D.; Peruzzi, L.; Cotti, R.; et al. New case of syncytial giant-cell variant of hepatocellular carcinoma in a pediatric patient with HNF1B deficiency: Does it fit with the syndrome? BMJ Open Gastroenterol. 2022, 9, e001013. [Google Scholar] [CrossRef] [PubMed]
- Gambella, A.; Mastracci, L.; Caporalini, C.; Francalanci, P.; Mescoli, C.; Ferro, J.; Alaggio, R.; Grillo, F. Not only a small liver—The pathologist’s perspective in the pediatric liver transplant setting. Pathologica 2022, 114, 89–103. [Google Scholar] [CrossRef]
- Kakos, C.D.; Ziogas, I.A.; Demiri, C.D.; Esagian, S.M.; Economopoulos, K.P.; Moris, D.; Tsoulfas, G.; Alexopoulos, S.P. Liver Transplantation for Pediatric Hepatocellular Carcinoma: A Systematic Review. Cancers 2022, 14, 1294. [Google Scholar] [CrossRef]
- Varol, F.I. Pediatric Hepatocellular Carcinoma. J. Gastrointest. Cancer 2020, 51, 1169–1175. [Google Scholar] [CrossRef]
- Torbenson, M.; Zen, Y.; Yeh, M.M. Tumors of the Liver: American Registry of Pathology; American Registry of Pathology: Rockville, MD, USA, 2018. [Google Scholar]
- Colclough, K.; Ellard, S.; Hattersley, A.; Patel, K. Syndromic Monogenic Diabetes Genes Should Be Tested in Patients with a Clinical Suspicion of Maturity-Onset Diabetes of the Young. Diabetes 2022, 71, 530–537. [Google Scholar] [CrossRef]
- Saint-Martin, C.; Bouvet, D.; Bastide, M.; Bellanne-Chantelot, C. Gene Panel Sequencing of Patients with Monogenic Diabetes Brings to Light Genes Typically Associated with Syndromic Presentations. Diabetes 2022, 71, 578–584. [Google Scholar] [CrossRef]
- Bingham, C.; Hattersley, A.T. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol. Dial. Transplant. 2004, 19, 2703–2708. [Google Scholar] [CrossRef]
- Adalat, S.; Bockenhauer, D.; Ledermann, S.E.; Hennekam, R.C.; Woolf, A.S. Renal malformations associated with mutations of developmental genes: Messages from the clinic. Pediatr. Nephrol. 2010, 25, 2247–2255. [Google Scholar] [CrossRef]
- Tudorache, E.; Sellier-Leclerc, A.L.; Lenoir, M.; Toubiana, N.; Bensman, A.; Bellanne-Chantelot, C.; Ulinski, T. Childhood onset diabetes posttransplant in a girl with TCF2 mutation. Pediatr. Diabetes 2012, 13, e35–e39. [Google Scholar] [CrossRef]
- Zuber, J.; Bellanné-Chantelot, C.; Carette, C.; Canaud, G.; Gobrecht, S.; Gaha, K.; Mallet, V.; Martinez, F.; Thervet, E.; Timsit, J.; et al. HNF1B-related diabetes triggered by renal transplantation. Nat Rev Nephrol. 2009, 5, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Teixeira, S. New-onset diabetes after kidney transplantation revealing HNF1B-associated disease. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Faguer, S.; Esposito, L.; Casemayou, A.; Pirson, Y.; Decramer, S.; Cartery, C.; Hazzan, M.; Garrigue, V.; Roussey, G.; Cointault, O.; et al. Calcineurin Inhibitors Downregulate HNF-1beta and May Affect the Outcome of HNF1B Patients After Renal Transplantation. Transplantation 2016, 100, 1970–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, E.R.; Badman, M.K.; Lockwood, C.R.; Clark, P.M.; Ellard, S.; Bingham, C.; Hattersley, A.T. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care 2004, 27, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Hattersley, A.T.; Greeley, S.A.; Polak, M.; Rubio-Cabezas, O.; Njølstad, P.R.; Mlynarski, W.; Castano, L.; Carlsson, A.; Raile, K.; Chi, D.V.; et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2018, 19 (Suppl. 27), 47–63. [Google Scholar] [CrossRef]
- Carrillo, E.; Lomas, A.; Pines, P.J.; Lamas, C. Long-lasting response to oral therapy in a young male with monogenic diabetes as part of HNF1B-related disease. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Haumaitre, C.; Barbacci, E.; Jenny, M.; Ott, M.O.; Gradwohl, G.; Cereghini, S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 1490–1495. [Google Scholar] [CrossRef] [Green Version]
- Maestro, M.A.; Boj, S.F.; Luco, R.F.; Pierreux, C.E.; Cabedo, J.; Servitja, J.M.; German, M.S.; Rousseau, G.G.; Lemaigre, F.P.; Ferrer, J. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum. Mol. Genet. 2003, 12, 3307–3314. [Google Scholar] [CrossRef] [Green Version]
- Haumaitre, C.; Fabre, M.; Cormier, S.; Baumann, C.; Delezoide, A.L.; Cereghini, S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum. Mol. Genet. 2006, 15, 2363–2375. [Google Scholar] [CrossRef] [Green Version]
- Haldorsen, I.S.; Vesterhus, M.; Raeder, H.; Jensen, D.K.; Søvik, O.; Molven, A.; Njølstad, P.R. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet. Med. 2008, 25, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Vesterhus, M.; Raeder, H.; Johansson, S.; Molven, A.; Njolstad, P.R. Pancreatic exocrine dysfunction in maturity-onset diabetes of the young type 3. Diabetes Care 2008, 31, 306–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edghill, E.L.; Oram, R.A.; Owens, M.; Stals, K.L.; Harries, L.W.; Hattersley, A.T.; Ellard, S.; Bingham, C. Hepatocyte nuclear factor-1beta gene deletions--a common cause of renal disease. Nephrol. Dial. Transplant. 2008, 23, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edghill, E.L.; Bingham, C.; Slingerland, A.S.; Minton, J.A.L.; Noordam, C.; Ellard, S.; Hattersley, A.T. Hepatocyte nuclear factor-1 beta mutations cause neonatal diabetes and intrauterine growth retardation: Support for a critical role of HNF-1beta in human pancreatic development. Diabet. Med. 2006, 23, 1301–1306. [Google Scholar] [CrossRef]
- Janky, R.S.; Binda, M.M.; Allemeersch, J.; Govaere, O.; Swinnen, J.V.; Roskams, T.; Aerts, S.; Topal, B. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer 2016, 16, 632. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kondratyeva, L.G.; Chernov, I.P.; Zinovyeva, M.V.; Kopantzev, E.P.; Sverdlov, E.D. Expression of master regulatory genes of embryonic development in pancreatic tumors. Dokl. Biochem. Biophys. 2017, 475, 250–252. [Google Scholar] [CrossRef]
- Kato, H.; Tateishi, K.; Fujiwara, H.; Nakatsuka, T.; Yamamoto, K.; Kudo, Y.; Hayakawa, Y.; Nakagawa, H.; Tanaka, Y.; Ijichi, H.; et al. MNX1-HNF1B Axis Is Indispensable for Intraductal Papillary Mucinous Neoplasm Lineages. Gastroenterology 2022, 162, 1272–1287 e16. [Google Scholar] [CrossRef]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Elliott, K.S.; Zeggini, E.; McCarthy, M.I.; Gudmundsson, J.; Sulem, P.; Stacey, S.N.; Thorlacius, S.; Amundadottir, L.; Grönberg, H.; Xu, J.; et al. Evaluation of association of HNF1B variants with diverse cancers: Collaborative analysis of data from 19 genome-wide association studies. PLoS ONE 2010, 5, e10858. [Google Scholar] [CrossRef]
- Thomson, E.; Tran, M.; Robevska, G.; Ayers, K.; van der Bergen, J.; Gopalakrishnan Bhaskaran, P.; Haan, E.; Cereghini, S.; Vash-Margita, A.; Margetts, M.; et al. Functional genomics analysis identifies loss of HNF1B function as a cause of Mayer-Rokitansky-Kuster-Hauser syndrome. Hum. Mol. Genet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.H.; Njolstad, P.R.; Horikawa, Y.; Bostad, L.; Bell, G.I.; Sovik, O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet. 1999, 8, 2001–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oram, R.A.; Edghill, E.L.; Blackman, J.; Taylor, M.J.; Kay, T.; Flanagan, S.E.; Ismail-Pratt, I.; Creighton, S.M.; Ellard, S.; Hattersley, A.T.; et al. Mutations in the hepatocyte nuclear factor-1beta (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am. J. Obstet. Gynecol. 2010, 203, 364 e1-5. [Google Scholar] [CrossRef] [PubMed]
- Haeri, S.; Devers, P.L.; Kaiser-Rogers, K.A.; Moylan, V.J.; Torchia, B.S.; Horton, A.L.; Wolfe, H.M.; Aylsworth, A.S. Deletion of hepatocyte nuclear factor-1-beta in an infant with prune belly syndrome. Am. J. Perinatol. 2010, 27, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Moreno-De-Luca, D.; Mulle, J.G.; Kaminsky, E.B.; Sanders, S.J.; Myers, S.M.; Adam, M.P.; Pakula, A.T.; Eisenhauer, N.J.; Uhas, K.; Weik, L.; et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet. 2010, 87, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loirat, C.; Bellanne-Chantelot, C.; Husson, I.; Deschenes, G.; Guigonis, V.; Chabane, N. Autism in three patients with cystic or hyperechogenic kidneys and chromosome 17q12 deletion. Nephrol. Dial. Transplant. 2010, 25, 3430–3433. [Google Scholar] [CrossRef]
- Nagamani, S.C.S.; Erez, A.; Shen, J.; Li, C.; Roeder, E.; Cox, S.; Karaviti, L.; Pearson, M.; Kang, S.H.L.; Sahoo, T.; et al. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet. 2010, 18, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Mefford, H.C.; Clauin, S.; Sharp, A.J.; Moller, R.S.; Ullmann, R.; Kapur, R.; Pinkel, D.; Cooper, G.M.; Ventura, M.; Ropers, H.H.; et al. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007, 81, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Quintero-Rivera, F.; Woo, J.S.; Bomberg, E.M.; Wallace, W.D.; Peredo, J.; Dipple, K.M. Duodenal atresia in 17q12 microdeletion including HNF1B: A new associated malformation in this syndrome. Am. J. Med. Genet. A 2014, 164, 3076–3082. [Google Scholar] [CrossRef]
- Murray, P.J.; Thomas, K.; Mulgrew, C.J.; Ellard, S.; Edghill, E.L.; Bingham, C. Whole gene deletion of the hepatocyte nuclear factor-1beta gene in a patient with the prune-belly syndrome. Nephrol. Dial. Transplant. 2008, 23, 2412–2415. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, N.W.; Clemens, M.; Canavan, T.P.; Surti, U.; Rajkovic, A. Prenatally diagnosed 17q12 microdeletion syndrome with a novel association with congenital diaphragmatic hernia. Fetal Diagn. Ther. 2012, 31, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Hinkes, B.; Hilgers, K.F.; Bolz, H.J.; Goppelt-Struebe, M.; Amann, K.; Nagl, S.; Bergmann, C.; Rascher, W.; Eckardt, K.U.; Jacobi, J. A complex microdeletion 17q12 phenotype in a patient with recurrent de novo membranous nephropathy. BMC Nephrol. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faguer, S.; Chassaing, N.; Bandin, F.; Prouheze, C.; Garnier, A.; Casemayou, A.; Huart, A.; Schanstra, J.P.; Calvas, P.; Decramer, S.; et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014, 86, 1007–1015. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patients with HNF1B Pathogenic Variants | Mean Age | Main Findings | Reference |
|---|---|---|---|
| 27 | 35 |
| [10] |
| 28 | 24 |
| [11] |
| 201 | >18 |
| [12] |
| 8 | 34.8 |
| [13] |
| 6 | 23 |
| [14] |
| 3 | 38.7 |
| [15] |
| 4 | 43.7 |
| [16] |
| Gender/Origin GW/BW g (DS) | Liver Involvement | Liver Histology | Renal Function and Ultrasound Findings | Pancreatic Involvement | Growth | Urogenital Malformations/ Cognitive Impairment | HNF1B Variant | Reference |
|---|---|---|---|---|---|---|---|---|
| ♂/Japan 39/2390 (−2.26) <1 month |
| PILBD, marked cholestasis |
| Diabetes requiring insulin therapy at 13 years of age (polyuria and polydipsia, mild metabolic acidosis) | NA | Absent/mild | c.457C>A, p.H135N (missense mutation in exon 2, de novo or paternal: history of liver dysfunction and renal insufficiency in his paternal family) | [63] |
| ♂/Belgium (Sardinian origin) 37/1520 (−3.46) <1 month |
| PILBD, severe biliary stasis, slight periportal fibrosis |
|
| Final height of 162.1 cm (−1.86 SD), BMI 19.0 kg/m2 (−0.62 SD) | Absent/NA | 499–504 delGCTCTG insCCCCT, A167FS (combination of a deletion and insertion in exon 2, de novo) | [62] |
| ♂/Germany 35/1780 (−1.69) <1 month |
| PILBD |
|
| Final height of 133.9 cm (−6.7 SD), BMI 17.3 kg/m2 (−2.1 SD) | Inguinal hernia, abdominal testis/delayed psychomotor development | HNF1B deletion exons 1–9, de novo | [61] |
| ♀/Czech Republic 38/2360 (−1.60) <1 month |
| PILBD, cholestasis without signs of bile duct proliferation |
|
| Growth along the 3rd centile | Absent/absent | 1698 kb deletion including HNF1B, de novo | [60] |
| ♂/France 35/NA <1 month |
|
|
| NA | NA | NA/NA | 1.5 Mb deletion including HNF1B | [64] |
| ♂/Italy 38/2600 (−1.27) <1 month |
| PILBD, biliary stasis |
| Initial pancreatic exocrine dysfunction without pancreatic hypoplasia at US | Growth along the 10th centile | Absent/absent | c.827G>A, p.R276Q (missense mutation in exon 4, de novo) | [65] |
| Gender/Origin GW/BW g (DS) Age of Presentation | Liver Involvement | Liver Histology | Renal Function and Ultrasound Findings | Pancreatic Involvement | Growth | Urogenital Malformations/ Cognitive Impairment | KIF12 Homozygous Variant (NM_138424.1) | Reference |
|---|---|---|---|---|---|---|---|---|
| (A) ♀/Syrian (consanguineous) full term 1 month |
| PILBD, bridging fibrosis with early nodule formation, mixed portal inflammatory infiltrate, multinucleated giant hepatocytes, extensive hepatocanalicular cholestasis, ductular reaction | Left hydronephrosis and mild increase in left renal pelvic anterior–posterior diameter to 6 mm | NA | NA | NA | c.655C>T: p. (Arg219 *) | [67] |
| (B) ♂/Turkish (consanguineous) full term 2 months |
| PILBD, biliary pattern of cirrhosis with nodule formation, mixed portal inflammatory infiltrate, mild ductular reaction, pseudo-acini formation, and nodule formation | Right renal pelvic anterior–posterior diameter | NA | NA | NA | c.610G>A: p. (Val204Met) | [67] |
| (C) ♂/Turkish (consanguineous) 9 years; ibling of patient B |
| Not performed | Caliectasis of the upper pole of the left kidney | NA | NA | NA | c.610G>A: p. (Val204Met) | [67] |
| (D) ♂/4 months |
| NA | Normal | Absent | Normal | Absent | c.463C>T: p. (Arg155*) | [68] |
| (E) ♂/Unknown (consanguineous)/5 years |
| NA | Normal | Absent | Normal | Absent | c.656G>A: p. (Arg219Gln) | [68] |
| (F) ♂/Unknown (consanguineous)/14 months |
| Suggestive of biliary cirrhosis | Normal | Absent | Normal | Absent | c.610G>A: p. (Val204Met) | [68] |
| (G) ♂/Unknown (consanguineous)/6 months |
| Suggestive of biliary atresia | Normal | Absent | Normal | Absent | c.610G>A: p. (Val204Met) | [68] |
| (E) ♀/Kurdish (consanguineous)/ 13 years |
| Extensive liver fibrosis, only minimal inflammation and proliferated bile ducts | Normal | Absent | Normal | Absent | c.655C>T: p. (Arg219 *) | [69] |
| (F) ♂/Kurdish (consanguineous)/ 13 years |
| Advanced fibrosis and bile duct proliferation | Normal | Pancreatic lipomatosis | Normal | Absent | c.655C>T: p. (Arg219 *) | [69] |
| (G) ♀/Iraqi (consanguineous)/ 7 years |
| NA | Normal | Absent | Normal | Absent | c.655C>T: p. (Arg219 *) | [69] |
| (H) ♀/Iraqi (consanguineous)/ 5 years |
| Canalicular cholestasis, moderate fibrosis, mild inflammation without steatosis | Normal | Absent | Failure to thrive | Absent | c.655C>T: p. (Arg219 *) | [69] |
| (I) ♂/Syrian (consanguineous)/12 years |
| Cirrhosis, septal hepatitis and ductular proliferation | Normal | Absent | Normal | Absent | c.655C>T: p. (Arg219 *) | [69] |
| (J) ♀/Afghan (consanguineous)/10 years |
| NA | Normal | Absent | Normal | Absent | c.482-4_500del p. ? | [69] |
| Genetic Disorders | Congenital Infections | Immune Disorders | Drug Related | |
|---|---|---|---|---|
| AGS (OMIM # 118450, OMIM # 610205) | HNF1B deficiency syndrome (OMIM # 137920) | Cytomegalovirus | Sclerosing cholangitis | Vanishing bile duct syndrome |
| Cystic fibrosis (OMIM # 219700) | KIF12-associated cholestasis (OMIM # 619662) | Rubella | Hemophagocytic lymphohistiocytosis | |
| α1-antitrypsin deficiency (OMIM # 613490) | ABBC12-associated cholestasis [88] | Syphilis | ||
| Niemann Pick type C (OMIM # 257220) | ||||
| Williams-Beuren syndrome (OMIM # 194050) | ||||
| Trisomy 21 (OMIM # 190685) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambella, A.; Kalantari, S.; Cadamuro, M.; Quaglia, M.; Delvecchio, M.; Fabris, L.; Pinon, M. The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored. Cells 2023, 12, 307. https://doi.org/10.3390/cells12020307
Gambella A, Kalantari S, Cadamuro M, Quaglia M, Delvecchio M, Fabris L, Pinon M. The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored. Cells. 2023; 12(2):307. https://doi.org/10.3390/cells12020307
Chicago/Turabian StyleGambella, Alessandro, Silvia Kalantari, Massimiliano Cadamuro, Marco Quaglia, Maurizio Delvecchio, Luca Fabris, and Michele Pinon. 2023. "The Landscape of HNF1B Deficiency: A Syndrome Not Yet Fully Explored" Cells 12, no. 2: 307. https://doi.org/10.3390/cells12020307