

Non-Steroidal Drug Interferences in a Quantitative Multisteroid LC-MS/MS Assay

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. LC-MS/MS Method
2.3. Screening for and Identification of Interferences
2.4. Confirmation of Found Interferences
3. Results
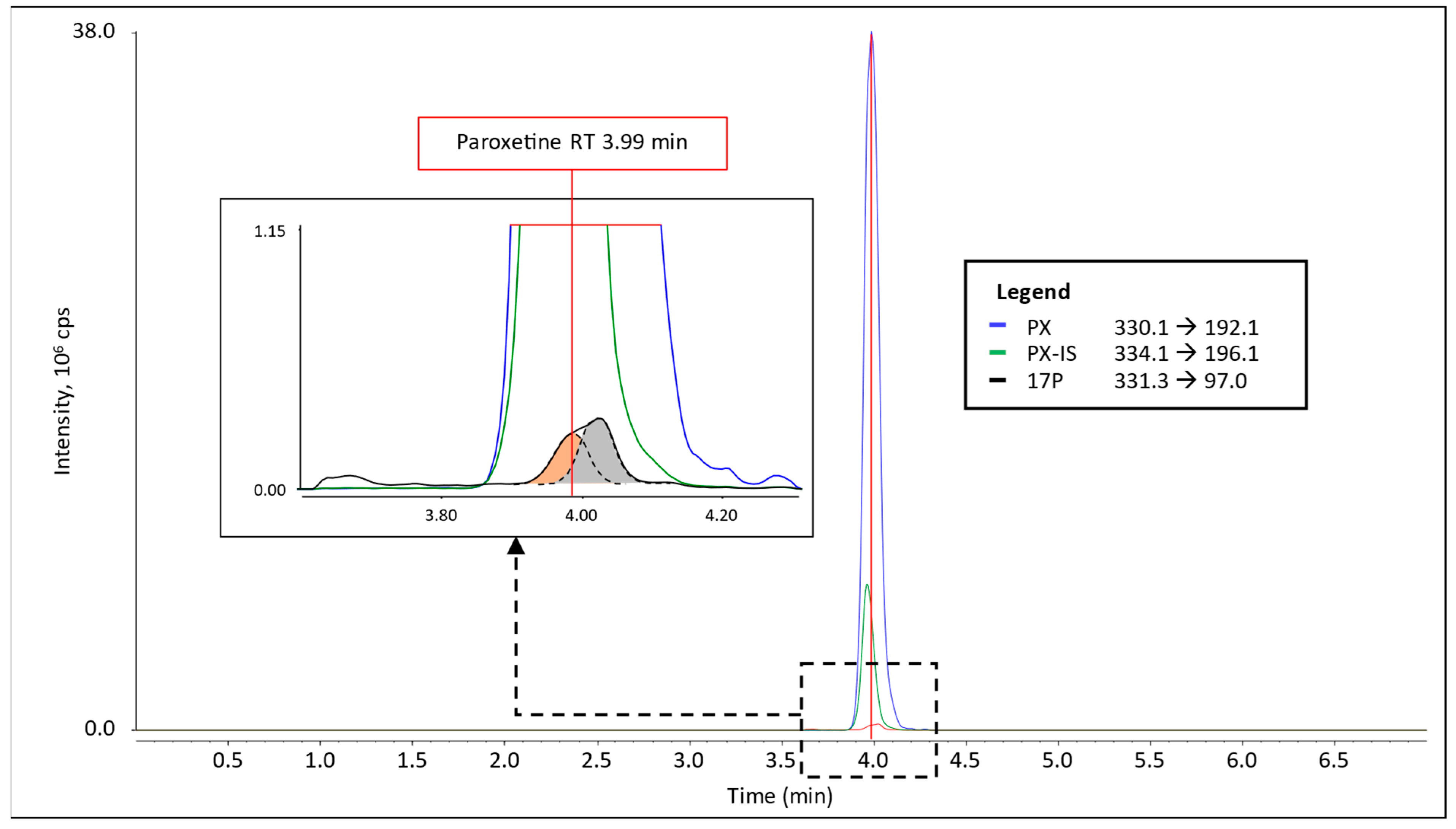
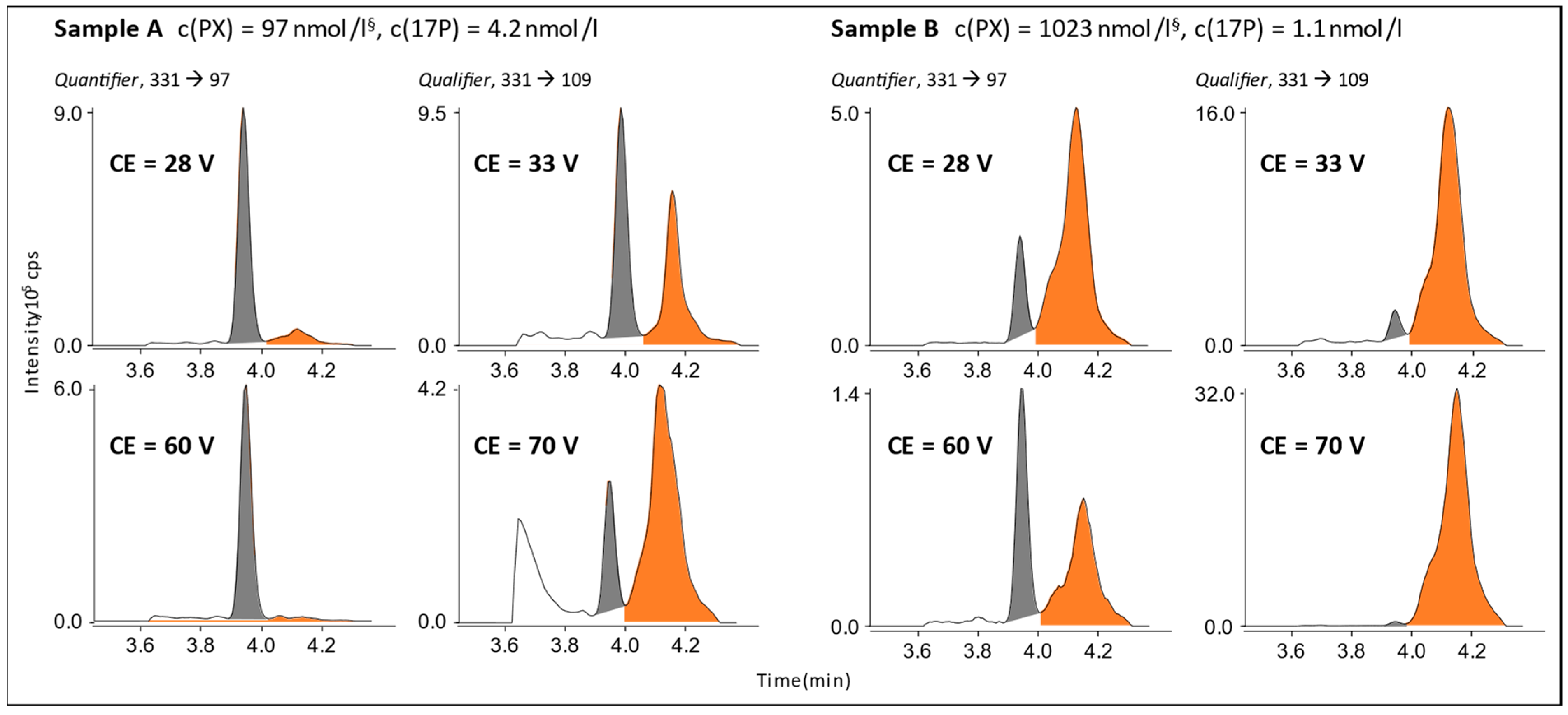
3.1. Interference in 17P Analysis
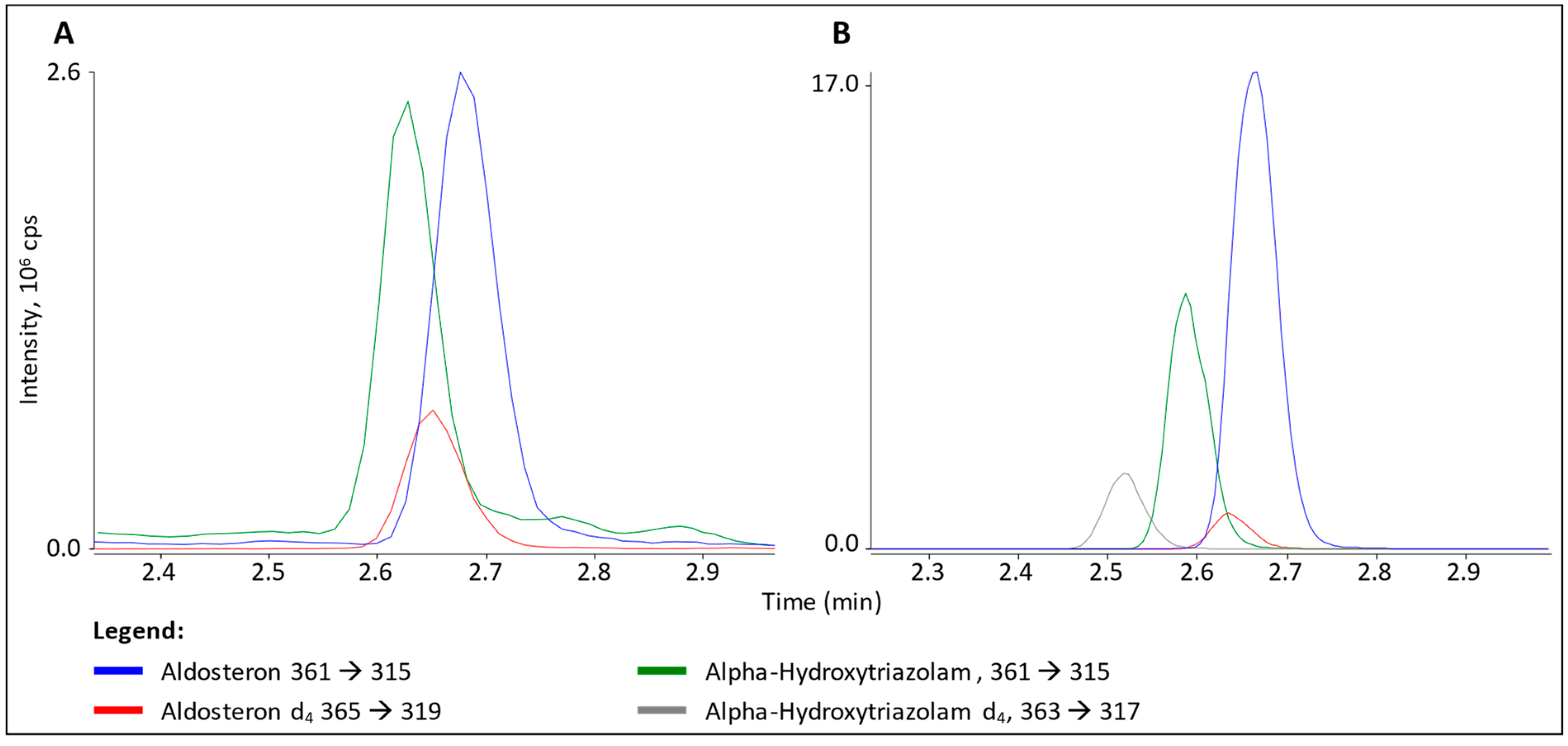
3.2. Interference in ALDO Analysis
4. Discussion
4.1. Paroxetine
4.2. α-Hydroxytriazolam
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seger, C.; Salzmann, L. After Another Decade: LC–MS/MS Became Routine in Clinical Diagnostics. Clin. Biochem. 2020, 82, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, M.M.; Rockwood, A.L.; Nelson, G.J.; Yue, B.; Urry, F.M. Assessing Analytical Specificity in Quantitative Analysis Using Tandem Mass Spectrometry. Clin. Biochem. 2005, 38, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Ionita, I.A.; Fast, D.M.; Akhlaghi, F. Development of a Sensitive and Selective Method for the Quantitative Analysis of Cortisol, Cortisone, Prednisolone and Prednisone in Human Plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven Golden Rules for Heuristic Filtering of Molecular Formulas Obtained by Accurate Mass Spectrometry. BMC Bioinformatics. 2007, 8, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drotleff, B.; Hallschmid, M.; Lämmerhofer, M. Quantification of Steroid Hormones in Plasma Using a Surrogate Calibrant Approach and UHPLC-ESI-QTOF-MS/MS with SWATH-Acquisition Combined with Untargeted Profiling. Anal. Chim. Acta 2018, 1022, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Stuppner, H.; Risch, L.; Seger, C. Design and Validation of a Sensitive Multisteroid LC-MS/MS Assay for the Routine Clinical Use: One-Step Sample Preparation with Phospholipid Removal and Comparison to Immunoassays. Int. J. Mol. Sci. 2022, 23, 14691. [Google Scholar] [CrossRef] [PubMed]
- Bagnoud, M.-A.; Reymond, J.-P. Interference of Metamizol (Dipyrone) on the Determination of Creatinine with the Kodak Dry Chemistry Slide Comparison with the Enzymatic Method from Boehringer. Clin. Chem. Lab. Med. 1993, 31, 753–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, J.M.; Griggs, D.A.; Nixon, A.L.; Long, W.H.; Flood, J.G. The Trazodone Metabolite Meta-Chlorophenylpiperazine Can Cause False-Positive Urine Amphetamine Immunoassay Results. J. Anal. Toxicol. 2011, 35, 364–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogeser, M.; Zachoval, R.; Spöhrer, U.; Jacob, K. Potential Lack of Specificity Using Electrospray Tandem-Mass Spectrometry for the Analysis of Mycophenolic Acid in Serum. Ther. Drug Monit. 2001, 23, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Seger, C.; Bock, M.; Tentschert, K.; Griesmacher, A. Evaluating Co-Medication and Matrix Interference Influence on Ion Yields: A Key Step in Therapeutic Drug Monitoring HPLC-MS/MS Assay Validation. Ann. Transplant. 2008, 13, 38. [Google Scholar]
- Chi, J.; Kim, M.; Li, F. Chromatography Quantitation of Paroxetine in Human Plasma by LC-MS/MS: Overcoming Significant Column-to-Column Retention Time Variation. J. Chromatogr. Sep. Tech. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Greaves, R.F.; Ho, C.S.; Loh, T.P.; Chai, J.H.; Jolly, L.; Graham, P.; Hartmann, M.F.; De Rijke, Y.B.; Wudy, S.A. Current State and Recommendations for Harmonization of Serum/Plasma 17-Hydroxyprogesterone Mass Spectrometry Methods. Clin. Chem. Lab. Med. 2018, 56, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration PAXIL® Presribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020031s060,020936s037,020710s024lbl.pdf (accessed on 14 December 2022).
- Jenkins-Jones, S.; Parviainen, L.; Porter, J.; Withe, M.; Whitaker, M.J.; Holden, S.E.; Morgan, C.L.; Currie, C.J.; Ross, R.J.M. Poor Compliance and Increased Mortality, Depression and Healthcare Costs in Patients with Congenital Adrenal Hyperplasia. Eur. J. Endocrinol. 2018, 178, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Wang, X.; Xu, L.; Wu, T.; Kang, D. Antidepressants for Polycystic Ovary Syndrome. Cochrane Database Syst. Rev. 2013, 2013, CD008575. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, D.; Rubin, K.H.; Nybo, M.; Abrahamsen, B.; Andersen, M. Morbidity and Medicine Prescriptions in a Nationwide Danish Population of Patients Diagnosed with Polycystic Ovary Syndrome. Eur. J. Endocrinol. 2015, 172, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, U.; Paff, D.; Klauber, W.L.J. Arznei Verordnungs—Report 2019; Springer: Berlin/Heidelberg, Germany, 2019; ISBN 9783662590454. [Google Scholar]
- Clincalc DrugStats Database—Antidepressants. Available online: https://clincalc.com/DrugStats/TC/Antidepressants (accessed on 6 December 2021).
- Norwegian Institute of Public Health (NIPH) Norwegian Prescription Database (NorPD). Available online: http://www.norpd.no/Prevalens.aspx (accessed on 8 December 2021).
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor-Semmes, R.L.; Kersey, K.; Williams, D.H.; Lam, R.; Koch, K.M. Effect of Ranitidine on the Pharmacokinetics of Triazolam and α-Hydroxytriazolam in Both Young (19–60 Years) and Older (61–78 Years) People. Clin. Pharmacol. Ther. 2001, 70, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Rush, C.R.; Baker, R.W.; Wright, K. Acute Behavioral Effects and Abuse Potential of Trazodone, Zolpidem and Triazolam in Humans. Psychopharmacology 1999, 144, 220–233. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, V.; Stuppner, H.; Seger, C. Non-Steroidal Drug Interferences in a Quantitative Multisteroid LC-MS/MS Assay. Cells 2023, 12, 329. https://doi.org/10.3390/cells12020329
Braun V, Stuppner H, Seger C. Non-Steroidal Drug Interferences in a Quantitative Multisteroid LC-MS/MS Assay. Cells. 2023; 12(2):329. https://doi.org/10.3390/cells12020329
Chicago/Turabian StyleBraun, Valentin, Hermann Stuppner, and Christoph Seger. 2023. "Non-Steroidal Drug Interferences in a Quantitative Multisteroid LC-MS/MS Assay" Cells 12, no. 2: 329. https://doi.org/10.3390/cells12020329