
Exploring the Relationship between Fusion Genes and MicroRNAs in Cancer

 ,
,  ,
,
Abstract
:1. Introduction
2. MiRNA in Precision Medicine and Precision Oncology
3. Fusion Genes and Chimeric Fusion Transcripts (Fusion RNAs)
4. Fusion Genes and MiRNAs
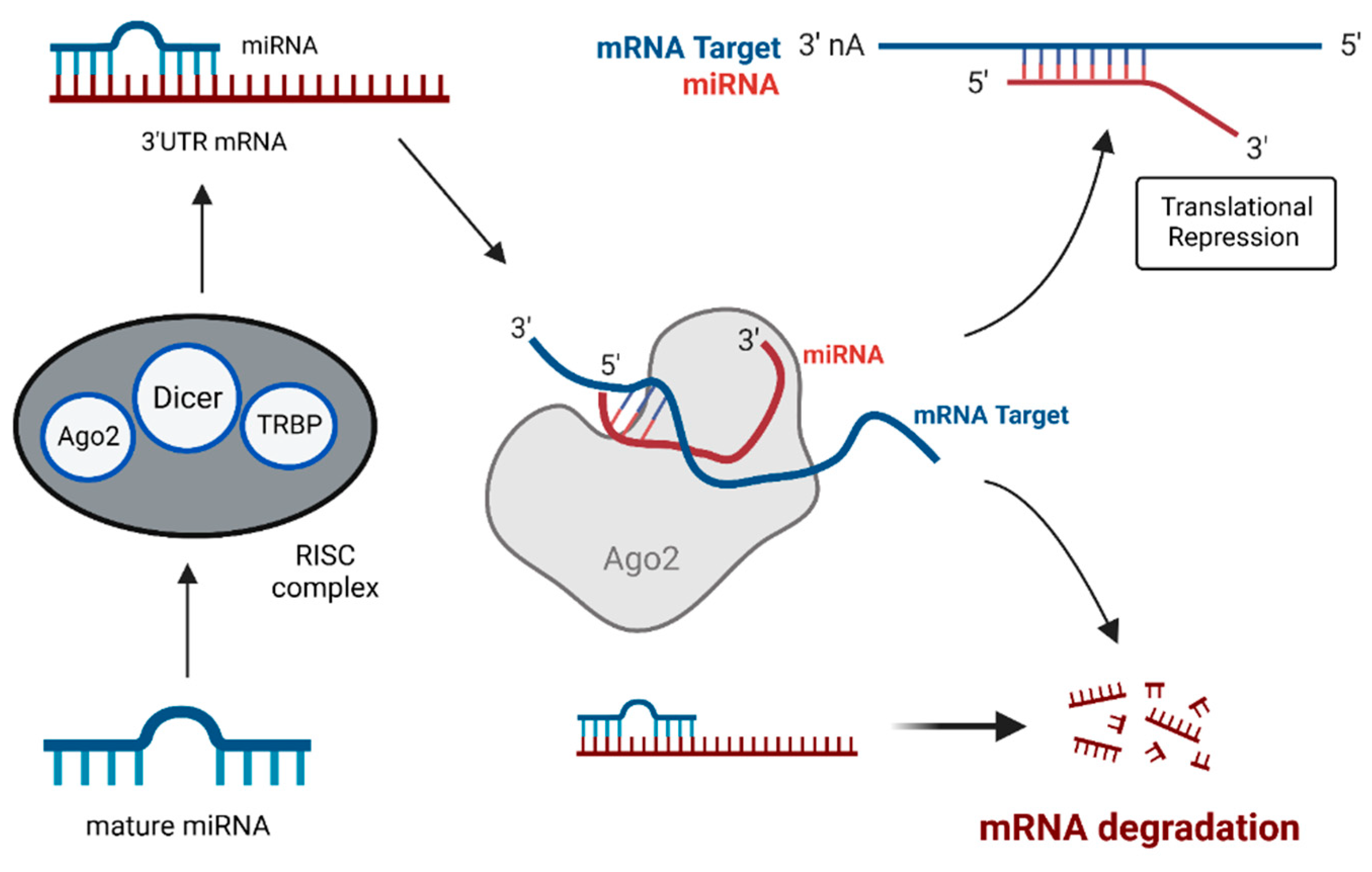
4.1. MiRNAs: Key Players in Gene Expression and Tumorigenesis
4.2. Mechanism of Fusion Gene-Mediated Aberrant miRNA Expression
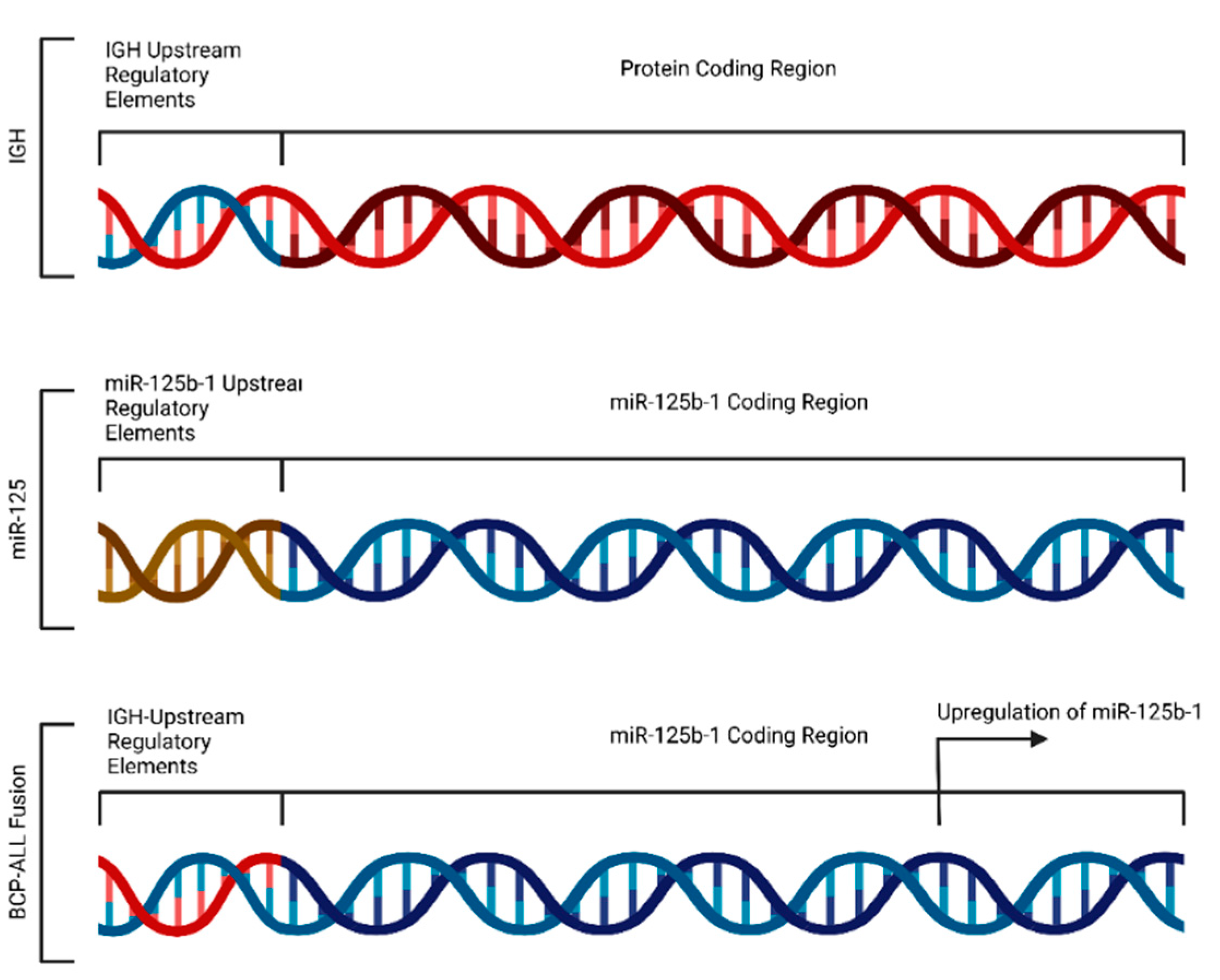
4.3. Fusion-Induced Promoter Transposition
4.4. Altered Expression of miR-29 in Lymphomas and Sarcomas
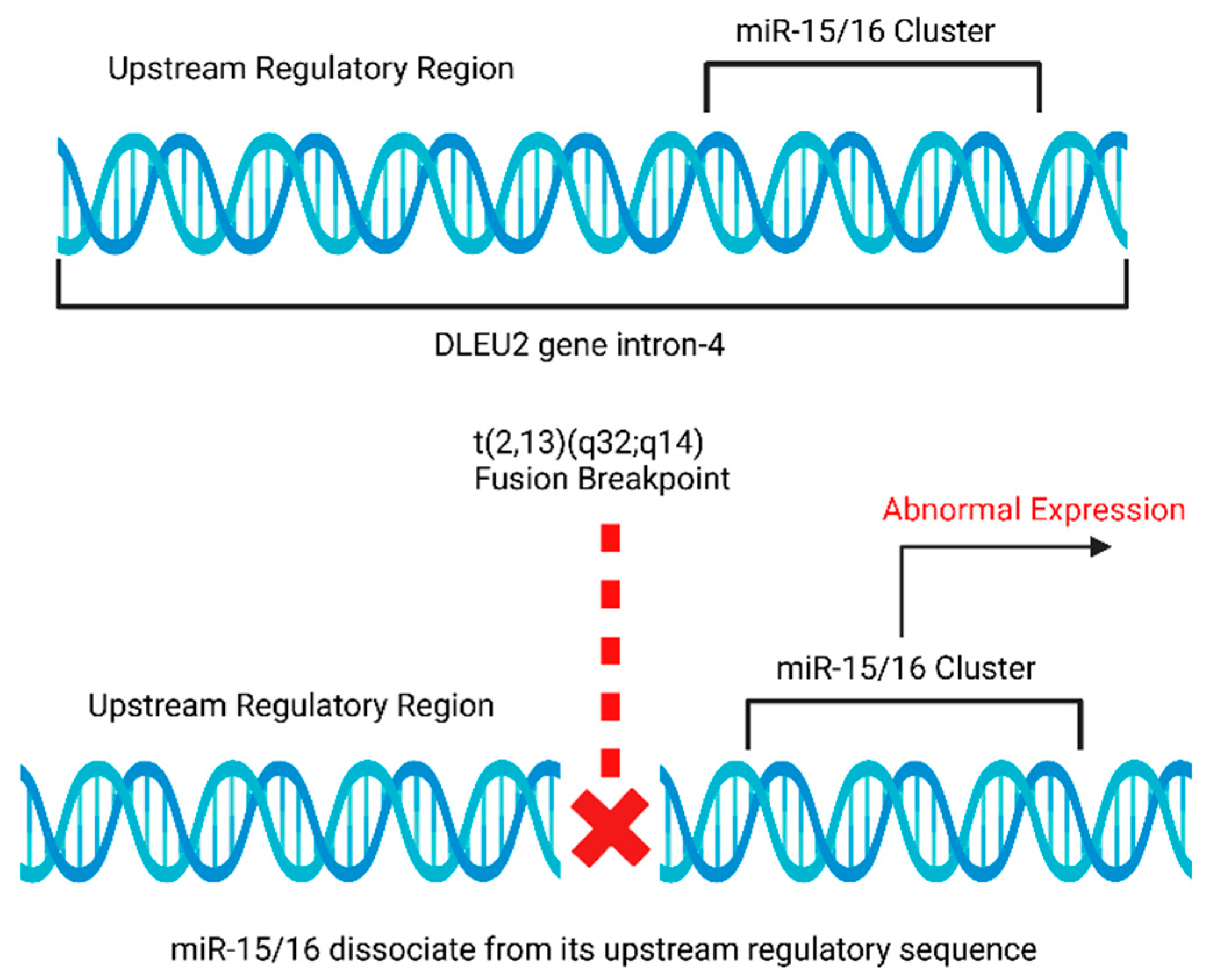
4.5. Rearrangements of miR-15a and miR-16-1 Loci in Chronic Lymphocytic Leukemia (CLL)
4.6. Translocation of miRNA-142 Causes c-MYC Overexpression in Acute Promyelocytic Leukemia (APL)
4.7. MiRNA Translocations in Mesenchymal Tumors and Benign Tumors
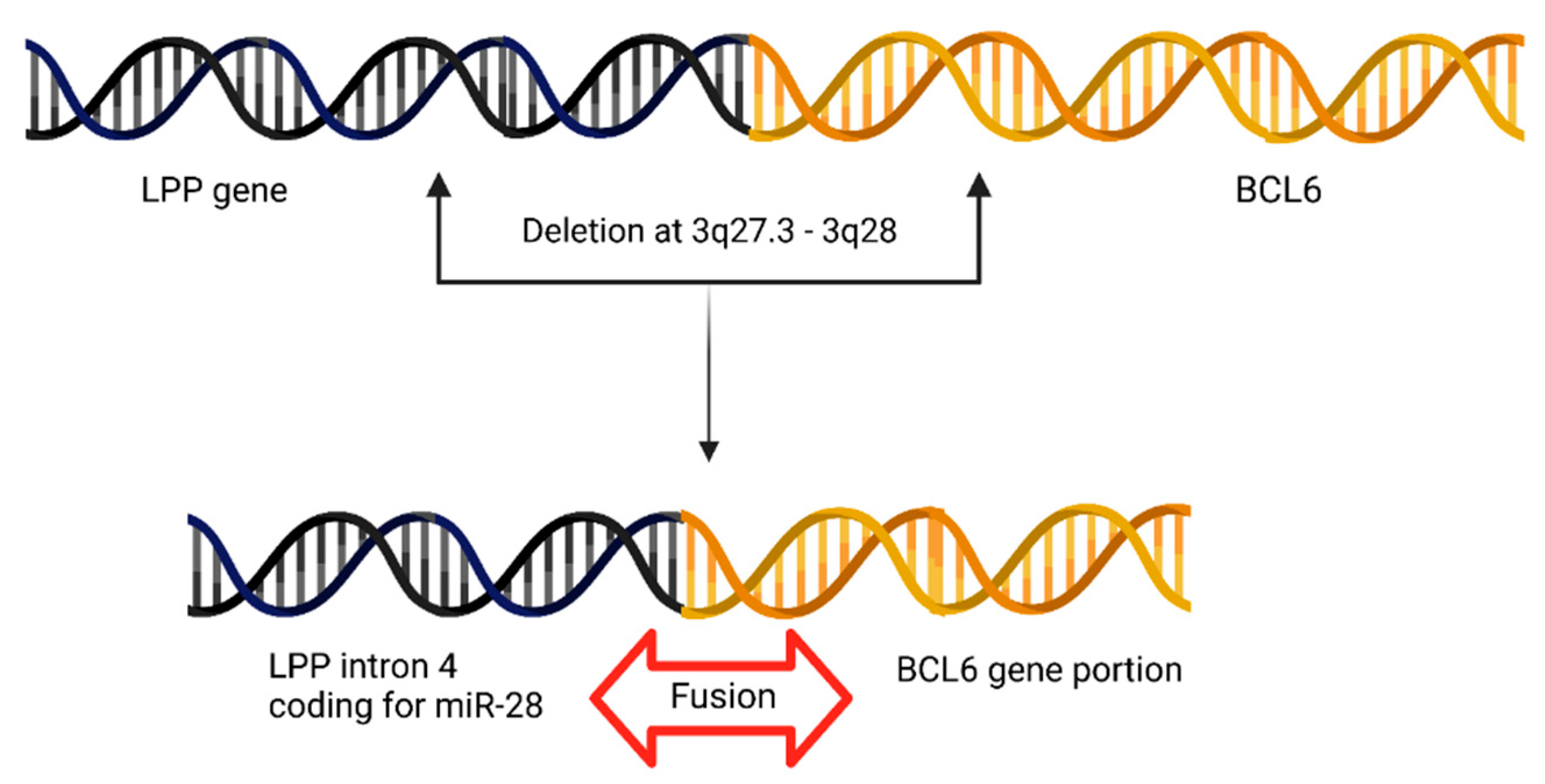
4.8. BCL6 Juxtapositions to miR-28 Locus in Primary Central Nervous System Lymphoma (PCNSL)
4.9. miR-17-92 Gene Cluster and Chronic Myeloid Leukemia (CML)
4.10. Fusion Genes Cause miRNA Overexpression in Solid Tumors
4.11. Abnormal miR-155 Levels in Burkitt’s Lymphoma Translocation
4.12. AML1-ETO Fusion Protein Causes miR-223 Repression
4.13. Clinical Relevance of Fusion Genes and miRNAs
5. MiRNA-Mediated Cancer Therapeutics in Fusion Positive Tumors
6. Latest Update: ChimerDriver Exploits miRNAs in Detection and Classification of Oncogenic Fusion Genes
7. Software and Databases to Identify miRNAs in Fusion Transcripts
8. Clinical Applications of Fusion Gene-miRNA Interplay
9. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tao, J.J.; Schram, A.M.; Hyman, D.M. Basket Studies: Redefining Clinical Trials in the Era of Genome-Driven Oncology. Annu. Rev. Med. 2018, 69, 319. [Google Scholar] [CrossRef] [PubMed]
- Panicker, S.; Venkatabalasubramanian, S.; Pathak, S.; Ramalingam, S. The Impact of Fusion Genes on Cancer Stem Cells and Drug Resistance. Mol. Cell Biochem. 2021, 476, 3771–3783. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Akimitsu, N. Fusion Genes and RNAs in Cancer Development. Noncoding RNA 2021, 7, 10. [Google Scholar] [CrossRef]
- Persson, H.; Søkilde, R.; Häkkinen, J.; Pirona, A.C.; Vallon-Christersson, J.; Kvist, A.; Mertens, F.; Borg, Å.; Mitelman, F.; Höglund, M.; et al. Frequent MiRNA-Convergent Fusion Gene Events in Breast Cancer. Nat. Commun. 2017, 8, 788. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Chandrashekar, D.S.; Varambally, S.; Creighton, C.J. Global Impact of Somatic Structural Variation on the Cancer Proteome. Nat. Commun. 2023, 14, 5637. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in Solid Tumours: Diagnostic Strategies, Targeted Therapy, and Acquired Resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Chapiro, E.; Russell, L.J.; Struski, S.; Cavé, H.; Radford-Weiss, I.; Valle, V.D.; Lachenaud, J.; Brousset, P.; Bernard, O.A.; Harrison, C.J.; et al. A New Recurrent Translocation t(11;14)(Q24;Q32) Involving IGH@ and MiR-125b-1 in B-Cell Progenitor Acute Lymphoblastic Leukemia. Leukemia 2010, 24, 1362–1364. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Özsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of Recurrent t(6;7)(P25.3;Q32.3) Translocations in ALK-Negative Anaplastic Large Cell Lymphomas by Massively Parallel Genomic Sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Loupe, J.M.; Miller, P.J.; Crabtree, J.S.; Zabaleta, J.; Hollenbach, A.D.; Loupe, J.M.; Miller, P.J.; Crabtree, J.S.; Zabaleta, J.; Hollenbach, A.D. Acquisition of an Oncogenic Fusion Protein Is Sufficient to Globally Alter the Landscape of MiRNA Expression to Inhibit Myogenic Differentiation. Oncotarget 2017, 8, 87054–87072. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Croce, C.M. Role of MiR-15/16 in CLL. Cell Death Differ. 2015, 22, 6. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and MiR-15/16: From Gene Discovery to Treatment. Cell Death Differ. 2017, 25, 21–26. [Google Scholar] [CrossRef]
- Gauwerky, C.E.; Huebner, K.; Isobe, M.; Nowell, P.C.; Croce, C.M. Activation of MYC in a Masked t(8;17) Translocation Results in an Aggressive B-Cell Leukemia. Proc. Natl. Acad. Sci. USA 1989, 86, 8867. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, K.; Enomoto, Y.; Suzuki, R.; Watanuki, J.; Hosoi, H.; Yamashita, Y.; Murata, S.; Mushino, T.; Tamura, S.; Hanaoka, N.; et al. Enforced Expression of MIR142, a Target of Chromosome Translocation in Human B-Cell Tumors, Results in B-Cell Depletion. Int. J. Hematol. 2018, 107, 345–354. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. Chromosomal Rearrangements and MicroRNAs: A New Cancer Link with Clinical Implications. J. Clin. Investig. 2007, 117, 2059. [Google Scholar] [CrossRef]
- Quade, B.J.; Weremowicz, S.; Neskey, D.M.; Vanni, R.; Ladd, C.; Cin, P.D.; Morton, C.C. Fusion Transcripts Involving HMGA2 Are Not a Common Molecular Mechanism in Uterine Leiomyomata with Rearrangements in 12q15. Cancer Res. 2003, 63, 1351–1358. [Google Scholar]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the Pairing between Let-7 and Hmga2 Enhances Oncogenic Transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Schwindt, H.; Akasaka, T.; Zühlke-Jenisch, R.; Hans, V.; Schaller, C.; Klapper, W.; Dyer, M.J.S.; Siebert, R.; Deckert, M. Chromosomal Translocations Fusing the BCL6 Gene to Different Partner Loci Are Recurrent in Primary Central Nervous System Lymphoma and May Be Associated with Aberrant Somatic Hypermutation or Defective Class Switch Recombination. J. Neuropathol. Exp. Neurol. 2006, 65, 776–782. [Google Scholar] [CrossRef]
- Venturini, L.; Battmer, K.; Castoldi, M.; Schultheis, B.; Hochhaus, A.; Muckenthaler, M.U.; Ganser, A.; Eder, M.; Scherr, M. Expression of the MiR-17-92 Polycistron in Chronic Myeloid Leukemia (CML) CD34+ Cells. Blood 2007, 109, 4399–4405. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A MicroRNA Expression Signature of Human Solid Tumors Defines Cancer Gene Targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Krichevsky, A.M.; Gabriely, G. MiR-21: A Small Multi-Faceted RNA. J. Cell Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, W.; Li, K.; Zheng, N.; Zheng, C.; Zheng, S. Identification of Hsa-MiR-21 as a Target Gene of Tumorigenesis in Neuroblastoma. Int. J. Clin. Exp. Med. 2016, 9, 2718–2727. [Google Scholar]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 Is an Antiapoptotic Factor in Human Glioblastoma Cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Schneider, C.; Setty, M.; Holmes, A.B.; Maute, R.L.; Leslie, C.S.; Mussolin, L.; Rosolen, A.; Dalla-Favera, R.; Bassoa, K. MicroRNA 28 Controls Cell Proliferation and Is Down-Regulated in B-Cell Lymphomas. Proc. Natl. Acad. Sci. USA 2014, 111, 8185–8190. [Google Scholar] [CrossRef]
- Liu, D.; Shimonov, J.; Primanneni, S.; Lai, Y.; Ahmed, T.; Seiter, K. T(8;14;18): A 3-Way Chromosome Translocation in Two Patients with Burkitt’s Lymphoma/Leukemia. Mol. Cancer 2007, 6, 35. [Google Scholar] [CrossRef]
- Fazi, F.; Nervi, C. MicroRNA: Basic Mechanisms and Transcriptional Regulatory Networks for Cell Fate Determination. Cardiovasc. Res. 2008, 79, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Letter: A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia Identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Parker, B.C.; Zhang, W. Fusion Genes in Solid Tumors: An Emerging Target for Cancer Diagnosis and Treatment. Chin. J. Cancer 2013, 32, 594. [Google Scholar] [CrossRef] [PubMed]
- Linardic, C.M. PAX3-FOXO1 Fusion Gene in Rhabdomyosarcoma. Cancer Lett. 2008, 270, 10. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 Gene Fusions in Bladder Cancer. Hum. Mol. Genet. 2013, 22, 795. [Google Scholar] [CrossRef]
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.; et al. The Tumorigenic FGFR3-TACC3 Gene Fusion Escapes MiR-99a Regulation in Glioblastoma. J. Clin. Invest. 2013, 123, 855–865. [Google Scholar] [CrossRef]
- Ou, S.H.I.; Horn, L.; Cruz, M.; Vafai, D.; Lovly, C.M.; Spradlin, A.; Williamson, M.J.; Dagogo-Jack, I.; Johnson, A.; Miller, V.A.; et al. Emergence of FGFR3-TACC3 Fusions as a Potential by-Pass Resistance Mechanism to EGFR Tyrosine Kinase Inhibitors in EGFR Mutated NSCLC Patients. Lung Cancer 2017, 111, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Deplus, R.; Delliaux, C.; Marchand, N.; Flourens, A.; Vanpouille, N.; Leroy, X.; Launoit, Y.d.; Duterque-Coquillaud, M. TMPRSS2-ERG Fusion Promotes Prostate Cancer Metastases in Bone. Oncotarget 2017, 8, 11827–11840. [Google Scholar] [CrossRef]
- Takezawa, K.; Okamoto, I.; Nishio, K.; Jänne, P.A.; Nakagawa, K. Role of ERK-BIM and STAT3-Survivin Signaling Pathways in ALK Inhibitor-Induced Apoptosis in EML4-ALK-Positive Lung Cancer. Clin. Cancer Res. 2011, 17, 2140–2148. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. Book. 2017, 37, 160–169. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular Profiling for Precision Cancer Therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.K.; Oluoha, O.; Patel, K.; Vanderwalde, A. Precision Medicine in Oncology: A Review of Multi-Tumor Actionable Molecular Targets with an Emphasis on Non-Small Cell Lung Cancer. J. Pers. Med. 2021, 11, 518. [Google Scholar] [CrossRef]
- Lin, V.T.G.; Yang, E.S. The Pros and Cons of Incorporating Transcriptomics in the Age of Precision Oncology. J. Natl. Cancer Inst. 2019, 111, 1016–1022. [Google Scholar] [CrossRef]
- Schee, K.; Lorenz, S.; Worren, M.M.; Günther, C.C.; Holden, M.; Hovig, E.; Fodstad, Ø.; Meza-Zepeda, L.A.; Flatmark, K. Deep Sequencing the MicroRNA Transcriptome in Colorectal Cancer. PLoS ONE 2013, 8, e66165. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.; Søkilde, R.; Häkkinen, J.; Vallon-Christersson, J.; Mitelman, F.; Borg, Å.; Höglund, M.; Rovira, C. Analysis of Fusion Transcripts Indicates Widespread Deregulation of SnoRNAs and Their Host Genes in Breast Cancer. Int. J. Cancer 2020, 146, 3343–3353. [Google Scholar] [CrossRef] [PubMed]
- Rovira, C. MiRNA-Convergent Gene Fusions. Mol. Cell Oncol. 2018, 5, e1406433. [Google Scholar] [CrossRef] [PubMed]
- Hafstað, V.; Søkilde, R.; Häkkinen, J.; Larsson, M.; Vallon-Christersson, J.; Rovira, C.; Persson, H. Regulatory Networks and 5′ Partner Usage of MiRNA Host Gene Fusions in Breast Cancer. Int. J. Cancer 2022, 151, 95–106. [Google Scholar] [CrossRef]
- Dai, X.; Theobard, R.; Cheng, H.; Xing, M.; Zhang, J. Fusion Genes: A Promising Tool Combating against Cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy Assessment of Fusion Transcript Detection via Read-Mapping and de Novo Fusion Transcript Assembly-Based Methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zou, Q.; Li, F.; Zhao, W.; Xu, H.; Zhang, W.; Deng, H.; Yang, X. Identification of the Cross-Strand Chimeric RNAs Generated by Fusions of Bi-Directional Transcripts. Nat. Commun. 2021, 12, 4645. [Google Scholar] [CrossRef]
- McCartney, A.M.; Hyland, E.M.; Cormican, P.; Moran, R.J.; Webb, A.E.; Lee, K.D.; Hernandez-Rodriguez, J.; Prado-Martinez, J.; Creevey, C.J.; Aspden, J.L.; et al. Gene Fusions Derived by Transcriptional Readthrough Are Driven by Segmental Duplication in Human. Genome Biol. Evol. 2019, 11, 2678. [Google Scholar] [CrossRef]
- Jividen, K.; Li, H. Chimeric RNAs Generated by Intergenic Splicing in Normal and Cancer Cells. Genes Chromosomes Cancer 2014, 53, 963–971. [Google Scholar] [CrossRef]
- Lu, B.; Jiang, R.; Xie, B.; Wu, W.; Zhao, Y. Fusion Genes in Gynecologic Tumors: The Occurrence, Molecular Mechanism and Prospect for Therapy. Cell Death Dis. 2021, 12, 783. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The Emerging Complexity of Gene Fusions in Cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Kaye, F.J. Mutation-Associated Fusion Cancer Genes in Solid Tumors. Mol. Cancer Ther. 2009, 8, 1399–1408. [Google Scholar] [CrossRef]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Cannell, I.G.; Kong, Y.W.; Bushell, M. How Do MicroRNAs Regulate Gene Expression? Biochem. Soc. Trans. 2008, 36, 1224–1231. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Felekkis, K.; Touvana, E.; Stefanou, C.; Deltas, C. MicroRNAs: A Newly Described Class of Encoded Molecules That Play a Role in Health and Disease. Hippokratia 2010, 14, 236. [Google Scholar]
- Russo, G.; Isobe, M.; Pegoraro, L.; Finan, J.; Nowell, P.C.; Croce, C.M. Molecular Analysis of a t(7;14)(Q35;Q32) Chromosome Translocation in a T Cell Leukemia of a Patient with Ataxia Telangiectasia. Cell 1988, 53, 137–144. [Google Scholar] [CrossRef]
- Lv, Y.; Yang, H.; Ma, X.; Wu, G. Strand-Specific MiR-28-3p and MiR-28-5p Have Differential Effects on Nasopharyngeal Cancer Cells Proliferation, Apoptosis, Migration and Invasion. Cancer Cell Int. 2019, 19, 187. [Google Scholar] [CrossRef]
- Tassano, E.; Acquila, M.; Tavella, E.; Micalizzi, C.; Panarello, C.; Morerio, C. MicroRNA-125b-1 and BLID Upregulation Resulting from a Novel IGH Translocation in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. Genes Chromosomes Cancer 2010, 49, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A Systematic Review of MiR-29 in Cancer. Mol. Ther. Oncolytics 2018, 12, 173–194. [Google Scholar] [CrossRef]
- Yano, M.; Toyooka, S.; Tsukuda, K.; Dote, H.; Morimoto, Y.; Ohata, N.; Ichimura, K.; Aoe, M.; Date, H.; Shimizu, N. SYT–SSX Fusion Genes in Synovial Sarcoma of the Thorax. Lung Cancer 2004, 44, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Thorson, J.A.; Weigelin, H.C.; Ruiz, R.E.; Howard, J.K.; Lucas, D.R. Identification of SYT-SSX Transcripts from Synovial Sarcomas Using RT-Multiplex PCR and Capillary Electrophoresis. Mod. Pathol. 2006, 19, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Salesse, S.; Verfaillie, C.M. BCR/ABL: From Molecular Mechanisms of Leukemia Induction to Treatment of Chronic Myelogenous Leukemia. Oncogene 2002, 21, 8547–8559. [Google Scholar] [CrossRef] [PubMed]
- de Braekeleer, M.; Guéganic, N.; Tous, C.; le Bris, M.J.; Basinko, A.; Morel, F.; Douet-Guilbert, N. Translocations Involving 13q14 without Associated Deletion in Chronic Lymphoid Leukaemia Target DLEU2. Br. J. Haematol. 2016, 172, 467–469. [Google Scholar] [CrossRef]
- Puiggros, A.; Blanco, G.; Espinet, B. Genetic Abnormalities in Chronic Lymphocytic Leukemia: Where We Are and Where We Go. Biomed. Res. Int. 2014, 2014, 435983. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. MiR-15 and MiR-16 Induce Apoptosis by Targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Hawthorn, L.A.; Chapman, L.R.; Oscier, D.; Cowell, J.K. The Consistent 13ql4 Translocation Breakpoint Seen in Chronic B-Cell Leukaemia (BCLL) Involves Deletion of the D13S25 Locus Which Lies Distal to the Retinoblastoma Predisposition Gene. Oncogene 1993, 8, 1415–1419. [Google Scholar] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 Family: Regulators of the Cellular Life-or-Death Switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A MicroRNA Polycistron as a Potential Human Oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Li, J.; Wan, Y.; Ji, Q.; Fang, Y.; Wu, Y. The Role of MicroRNAs in B-Cell Development and Function. Cell Mol. Immunol. 2013, 10, 107–112. [Google Scholar] [CrossRef]
- de Yébenes, V.G.; Bartolomé-Izquierdo, N.; Ramiro, A.R. Regulation of B-Cell Development and Function by MicroRNAs. Immunol. Rev. 2013, 253, 25–39. [Google Scholar] [CrossRef]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B Cell Proliferation and Lymphoblastic Leukemia/High-Grade Lymphoma in E(Mu)-MiR155 Transgenic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef]
- Metzler, M.; Wilda, M.; Busch, K.; Viehmann, S.; Borkhardt, A. High Expression of Precursor MicroRNA-155/BIC RNA in Children with Burkitt Lymphoma. Genes Chromosomes Cancer 2004, 39, 167–169. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNAs and Chromosomal Abnormalities in Cancer Cells. Oncogene 2006, 25, 6202–6210. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA Signatures in Human Cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Søkilde, R.; Persson, H.; Ehinger, A.; Pirona, A.C.; Fernö, M.; Hegardt, C.; Larsson, C.; Loman, N.; Malmberg, M.; Rydén, L.; et al. Refinement of Breast Cancer Molecular Classification by MiRNA Expression Profiles. BMC Genom. 2019, 20, 503. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Richards, R.I. Fragile and Unstable Chromosomes in Cancer: Causes and Consequences. Trends Genet. 2001, 17, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-MiRNA Oligonucleotides: A Comprehensive Guide for Design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef]
- Esau, C.C. Inhibition of MicroRNA with Antisense Oligonucleotides. Methods 2008, 44, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, S.; Patutina, O. Enhanced Inhibition of Tumorigenesis Using Combinations of MiRNA-Targeted Therapeutics. Front. Pharmacol. 2019, 10, 488. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, R.; Hussain, M.U. MicroRNAs and Human Diseases: Diagnostic and Therapeutic Potential. Cell Tissue Res. 2014, 358, 1–15. [Google Scholar] [CrossRef]
- Tonouchi, E.; Gen, Y.; Muramatsu, T.; Hiramoto, H.; Tanimoto, K.; Inoue, J.; Inazawa, J. MiR-3140 Suppresses Tumor Cell Growth by Targeting BRD4 via Its Coding Sequence and Downregulates the BRD4-NUT Fusion Oncoprotein. Sci. Rep. 2018, 8, 4482. [Google Scholar] [CrossRef]
- Tian, X.M.; Luo, Y.Z.; He, P.; Li, J.; Ma, Z.W.; An, Y. Inhibition of Invasion and Migration of Prostate Cancer Cells by MiRNA-509-5p via Targeting MDM2. Genet. Mol. Res. 2017, 16, gmr16019195. [Google Scholar] [CrossRef]
- Lovino, M.; Montemurro, M.; Barrese, V.S.; Ficarra, E. Identifying the Oncogenic Potential of Gene Fusions Exploiting MiRNAs. J. Biomed. Inform. 2022, 129, 104057. [Google Scholar] [CrossRef]
- Abate, F.; Zairis, S.; Ficarra, E.; Acquaviva, A.; Wiggins, C.H.; Frattini, V.; Lasorella, A.; Iavarone, A.; Inghirami, G.; Rabadan, R. Pegasus: A Comprehensive Annotation and Prediction Tool for Detection of Driver Gene Fusions in Cancer. BMC Syst. Biol. 2014, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Lovino, M.; Ciaburri, M.S.; Urgese, G.; di Cataldo, S.; Ficarra, E. DEEPrior: A Deep Learning Tool for the Prioritization of Gene Fusions. Bioinformatics 2020, 36, 3248–3250. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017, 120295. [Google Scholar] [CrossRef]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based MiRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The MicroRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Shepherd, C.J.; Aladowicz, E.; Olmos, D.; Selfe, J.; Pierron, G.; Delattre, O.; Walters, Z.; Shipley, J. MicroRNA and Gene Co-Expression Networks Characterize Biological and Clinical Behavior of Rhabdomyosarcomas. Cancer Lett. 2017, 385, 251–260. [Google Scholar] [CrossRef] [PubMed]



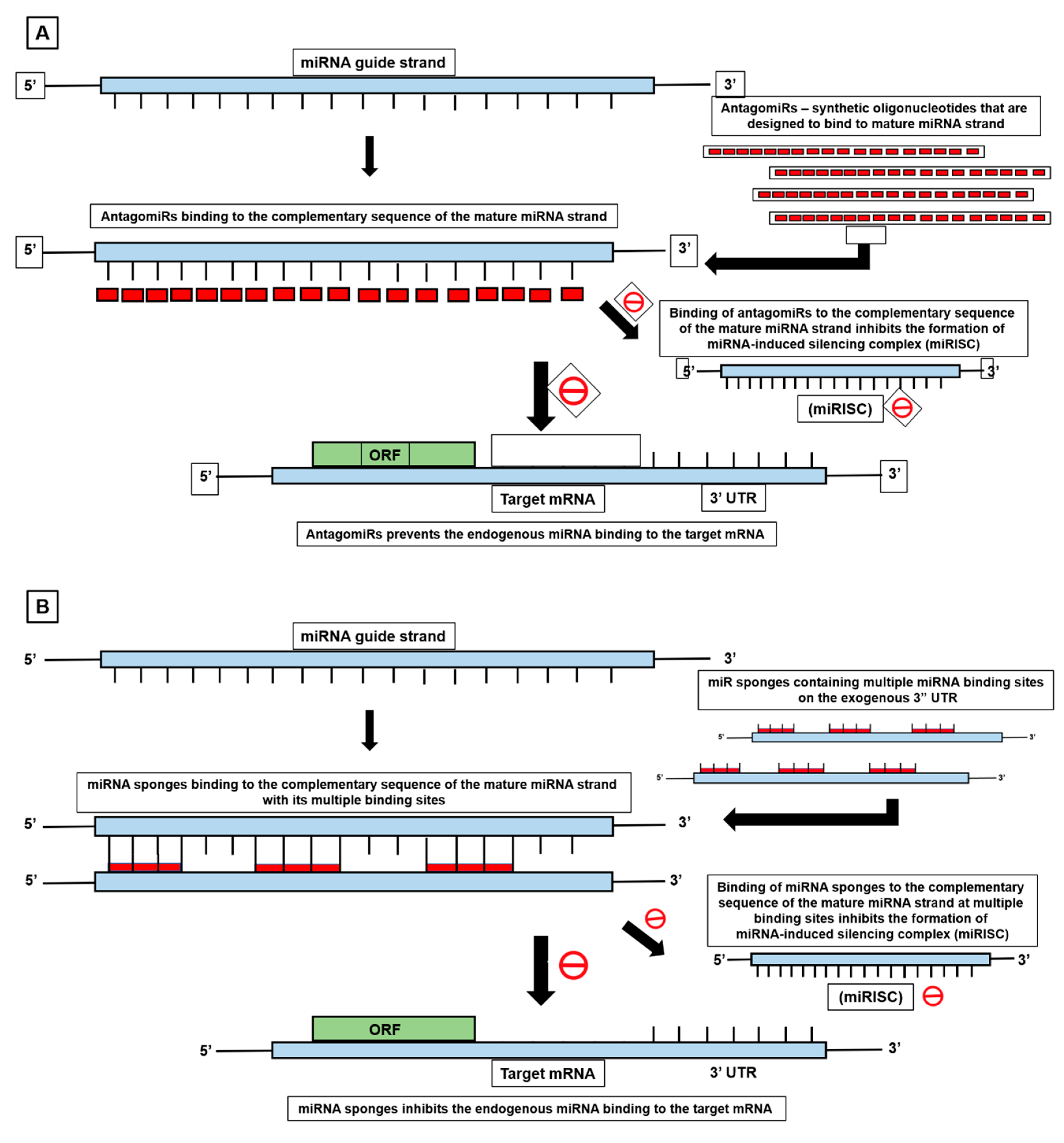


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Fusion Gene | Dysregulated miRNA | Cancer Type | Reference |
|---|---|---|---|---|
| 1 | BCP-ALL | miR-125b-1 | Myeloid cancer | [7] |
| 2 | * Translocation at 7q32.3 | miR-29 | ALK-negative large cell lymphoma | [8] |
| 3 | PAX-FOXO1 | miR-29-a-3p | Alveolar rhabdomyosarcoma | [9] |
| 4 | * Deletion at 13q14 | miR-15/16 | Chronic lymphocytic leukemia | [10,11] |
| 5 | * t(8;17)(q24;q22) | miR-142 | Acute promyelocytic leukemia | [12,13] |
| 6 | * Translocation at 12q15 | Let-7 miRNA | Mesenchymal and benign tumors | [14,15,16] |
| 7 | BCL6-LPP | miR-28 | Primary Central Nervous System Lymphoma | [17] |
| 8 | BCL-ABL | miR-17-92 | Chronic myeloid leukemia | [14,18] |
| 9 | * Translocation at 17q23.2 | miR-21 | Glioblastoma | [14,19,20,21,22] |
| 10 | * t(8;14)(q24;q32) | miR-155 | B-cell lymphoma | [23,24] |
| 11 | AML1-ETO | miR-223 | Acute myeloid leukemia | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panicker, S.; Chengizkhan, G.; Gor, R.; Ramachandran, I.; Ramalingam, S. Exploring the Relationship between Fusion Genes and MicroRNAs in Cancer. Cells 2023, 12, 2467. https://doi.org/10.3390/cells12202467
Panicker S, Chengizkhan G, Gor R, Ramachandran I, Ramalingam S. Exploring the Relationship between Fusion Genes and MicroRNAs in Cancer. Cells. 2023; 12(20):2467. https://doi.org/10.3390/cells12202467
Chicago/Turabian StylePanicker, Saurav, Gautham Chengizkhan, Ravi Gor, Ilangovan Ramachandran, and Satish Ramalingam. 2023. "Exploring the Relationship between Fusion Genes and MicroRNAs in Cancer" Cells 12, no. 20: 2467. https://doi.org/10.3390/cells12202467