

Role of Actin-Binding Proteins in Skeletal Myogenesis




Abstract
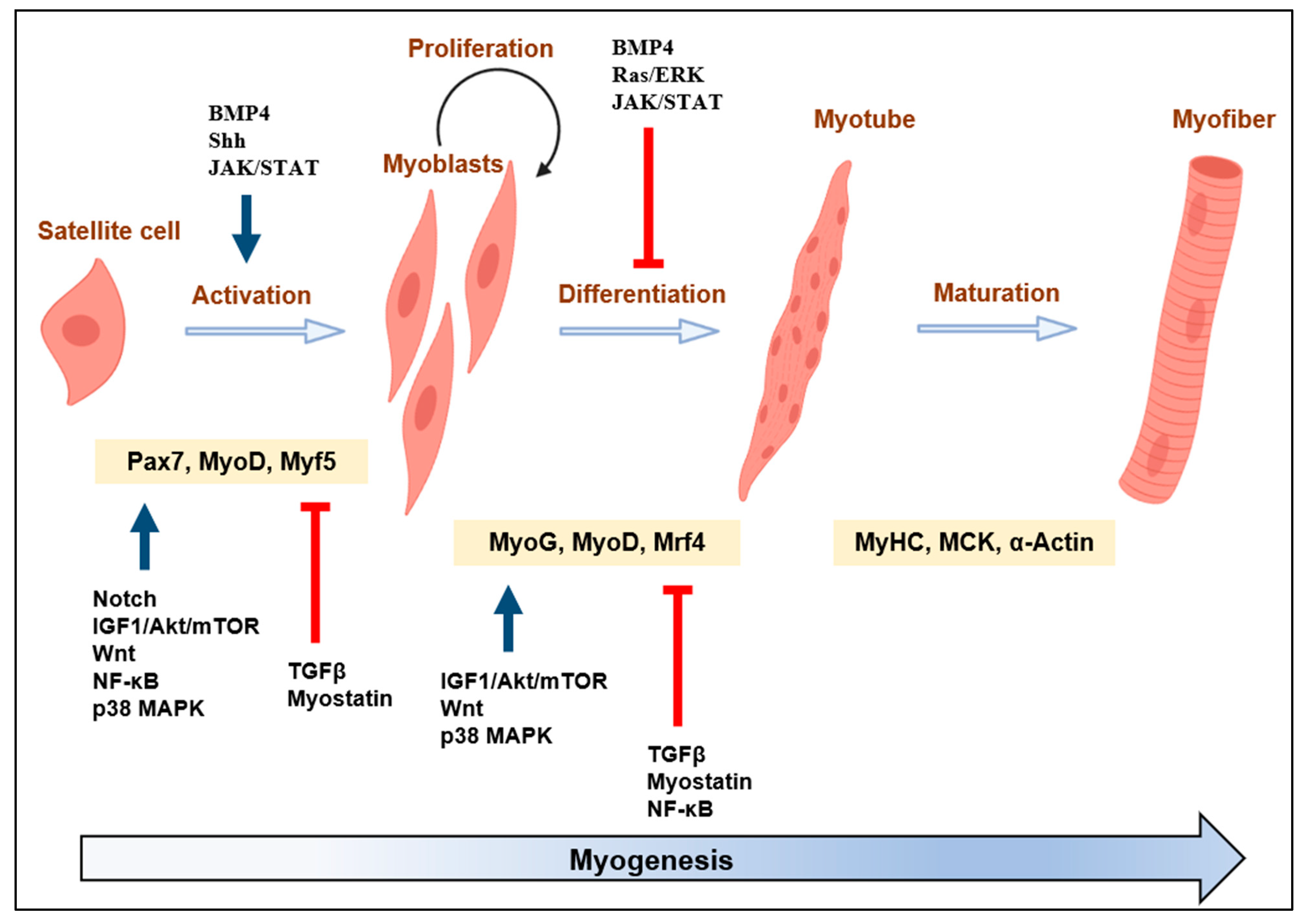
:1. Introduction
2. Overview of Actin Cytoskeleton Dynamics, ABPs, and Myogenesis

{kind=link}
{kind=link}
{kind=link}
| Protein Group | Actin-Binding Protein | Main Function | Refs. |
|---|---|---|---|
| Proteins regulating F-actin assembly | Profilin (PFN) | Binds to and sequesters actin monomer and promotes actin polymerization. | [37,38] |
| Proteins for F-actin dissambly | ADF/cofilin family (Destrin (Des), Cofilin (CFL)) | Bind both F-actin and G-actin, leading to depolymerization from the pointed ends | [39,40] |
| WD-repeat domain 1 (WDR1) | Major cofactor of ADF/CFL | [41,42] | |
| Twinfilin (TWF) | Binds and sequesters ADP-actin monomer, blocks filament elongation, and caps activity | [43,44,45] | |
| Cyclase-associated protein (CAP) | Required to activate adenylyl cyclase, which binds to G and F actin, ADF/CFL partner, regulating actin filament dynamics | [46,47] | |
| Monomer-sequestering proteins | Thymosinb4 (Tb4) | Sequesters actin monomer, and prevent it from engaging in the polymerization reactions | [25] |
| Myocardin-Related Transcription Factor (MRTF-A) | Controls G-actin/F-actin balance, generating a closed link between actin dynamics and gene expression. | [48] | |
| Proteins for actin nucleation | Formins | Enables the elongation of actin filament at the barded end | [49] |
| Proteins for nucleation sites in actin branching | Actin-related protein-2/3 (Arp2/3) | Promotes actin nucleation to generate a new branched (daughter) actin network by binding to the side of the existing filament (mother) | [50,51] |
| Wiskott—Aldrich syndrome protein (WASP), suppressor of cyclic AMP repressor (SCAR or WAVE) | Activate Arp2/3 complex to induce the branching of a new filament | [52,53] | |
| S. cerevisiae actin-binding protein-1 (Abp1), Pan1, and Cortactin (CTTN) | Promote and stabilize Arp2/3-mediated branches | [50,54] | |
| Capping proteins | CapZ | Binds barbed ends to stop filament growth | [55,56] |
| Gelsolin (Gel) | Promotes the nucleation step of actin polymerization | [57] | |
| Tropomodulin (Tmod)/Leiomodin (Lmod) | Cap the pointed ends of the actin-based thin filaments in striated muscle | [58] | |
| Actin filament cross-linking proteins | Fascin (FSCN) | Promotes filopodia assembly, and stable actin bundles | [59] |
| Filamin (Fln) | Binds F-actin into orthogonal branches, cross-link F-actin | [60] | |
| F-actin stabilizing proteins | Calponin (CNN) | Inhibit actomyosin ATPase, regulate and stabilize actin filament motility | [61] |
| Drebrin (Dbn) | Binds to the F-actin side and promotes F-actin formation | [62] | |
| Microtubule associated monooxygenase (MICAL) | Induces redox reactions on F-actin, makes certain disaggregation, and prevents polymerization | [63,64] | |
| Nexilin (Nelin, NEXN) | F-actin cross-linking activity through binding along the sides of F-actin | [65] | |
| XIN-repeating protein (XIN) | Prevents depolymerization by binding to F-actin | [66] | |
| ABPs related to muscle contraction | α-actinin, myosin IIs (NM IIs), Nebulin, Tropomyosins (Tpms) | Stabilize and regulate F-actin | [67,68,69,70,71,72] |
3. Roles of ABPs on Actin Remodeling and Skeletal Myogenesis
3.1. Proteins Regulating F-Actin Assembly
Profilin (PFN)
3.2. Proteins Regulating F-Actin Disassembly
3.2.1. Actin-Depolymerizing Factor/Cofilin Family Proteins (ADF/Cofilin)
3.2.2. WD-Repeat Protein 1 (WDR1)
3.2.3. Cyclase-Associated Actin Cytoskeleton Regulatory Protein (CAP)
3.2.4. Twinfilin (TWF)
3.3. Monomer-Sequestering Proteins
3.3.1. Thymosin β4 (Tβ4)
3.3.2. Myocardin-Related Transcription Factor (MRTF)
3.4. Proteins for Nucleation Sites in Actin Branching
3.4.1. Actin-Related Protein-2/3 (Arp2/3) and WASP/WAVE
3.4.2. Formin Homology Domains (FHOD)
3.4.3. Disheveled-Associated Activator of Morphogenesis (DAAM)
3.4.4. Protein Diaphanous (Dia)
3.4.5. Cortactin (CTTN)
3.5. Capping Proteins
3.5.1. Gelsolin (Gel)
3.5.2. F-Actin-Capping Protein Subunit Beta 1 (CapZβ1)
3.5.3. Tropomodulins (Tmods)
3.5.4. Leiomodin (Lmod)
3.6. Actin Filament Cross-Linking Proteins
3.6.1. Filamins (Flns)
3.6.2. Fascin
3.7. F-Actin Stabilizing Proteins
3.7.1. Calponin (CNN)
3.7.2. Drebrin (Dbn1)
3.7.3. Xin Actin-Binding Repeat-Containing Protein 1 (Xin)
3.7.4. Nexilin (NEXN)
3.7.5. [F-Actin]-Monooxygenase MICAL2
3.8. Muscle Contractile-Related ABPs in Myogenic Differentiation
3.8.1. α-Actinin
3.8.2. Nebulin
4. Epigenetic Regulation of ABPs by ncRNAs in Skeletal Myogenesis
| lncRNAs/miRNAs | Functions | Mechanisms | Refs. |
|---|---|---|---|
| lnc23 | Promotes myogenic differentiation | Lnc23 may release RhoA and Rac1 from the inhibitory effect of PFN1 by binding to PFN1 | [250] |
| miR-1/miR-133 | Induces actin disorganization during sarcomere assembly | Target PFN2, Arp2/3 | [251] |
| miR-1 | Negatively regulates skeletal muscle development | Targets CNN3 | [212] |
| miR-206 | Glycolysis regulator during prenatal skeletal muscle development | [212,253] | |
| miR-182 | Promotes bovine primary myoblast differentiation | Targets CFL1 | [89] |
| miR-204 | Improved in myocardial ischemia/reperfusion injury in mice | [258] | |
| miR-320-3p | Negatively regulates myogenic differentiation | Targets CFL2 | [259] |
| miR-141-3p | Negatively regulates myogenic differentiation | [262] | |
| miR-325-3p | Negatively regulates myogenic differentiation | [261] | |
| miR-429-3p | Negatively regulates myogenic differentiation | [260] | |
| miR-145 | Increases stress fiber formation by modulating actin dynamics and cytoskeletal assembly | [271] | |
| miR-1/133/206 | Promotes myogenic differentiation | Targets CAP1 | [100] |
| miR-1/206miR-486 | Promotes myogenic differentiation | MRTF-A | [192] |
| lncPRRX1/ miR-137 | Promotes myogenic differentiation | lncPRRX1 repaired the defects in Cdc42 protein levels caused by miR-137 | [270] |
| miR-320 | Promotes myoblasts differentiation | Targets Grb2 | [264] |
| miR-200a | Promotes cell differentiation and proliferation | [265] |
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABPs | Actin-binding proteins |
| Abi | Abl interactor 1 also known as Abelson interactor 1 |
| Abp1 | S. cerevisiae actin-binding protein-1 |
| ADF | Actin depolymerization factor |
| Akt | Serine/threonine kinase |
| AMPK | AMP-activated protein kinase |
| Arp2/3 | Actin-related protein-2/3 |
| BMP | Bone Morphogenetic Proteins |
| CAP | Cyclase-associated protein |
| CapZ | Capping protein |
| Cdc42 | Cell Division Cycle 42 |
| CFL | Cofilin |
| CNN | Calponin |
| CTTN | Cortactin |
| Dbn | Drebrin |
| Des | Destrin |
| Dvl2 | Disheveled-2 |
| EB3 | End-binding 1 |
| EGFR | Epidermal growth factor receptor |
| EVH1 | Ena/VASP homology 1 |
| Fln | Filamin |
| Fscn | Fascin |
| Gel | Gelsolin |
| Grb2 | Growth factor receptor-bound protein 2 |
| HDAC1 | Histone Deacetylase 1 |
| HSPB7 | Heat Shock Protein Family B member 7 |
| HSPC300 | Haematopoietic stem/progenitor cell protein 300 |
| IGF-1 | Insulin-like growth factor 1 |
| IRS1 | Insulin receptor substrate 1 |
| IRSp53 | Insulin receptor substrate protein 53kDa |
| JAK/STAR | Janus kinase/signal transducer and activator of transcription |
| LncRNAs | Long non-coding RNAs |
| Lmod | Leiomodin |
| MCK | Muscle creatine kinase |
| MHC | Myosin heavy chain |
| MICAL | Microtubule Associated Monooxygenasep |
| MicroRNAs | miRNAs |
| Mrf4 | Myogenic regulatory factor 4 |
| MRFs | Myogenic regulatory factors |
| mTOR | Mammalian target of rapamycin |
| Myf5 | Myogenic factor 5 |
| MyoD | Myogenic factor 3 |
| MyoG | Myogenin |
| Nap1/Hem2/Kette | Nck-associated protein 1/Hematopoietic protein 2/Kette |
| Nck | Non-catalytic region of tyrosine kinase |
| ncRNAs | Non-coding RNAs |
| NEXN | Nexilin, Nelin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| N-WASP | Neural- WASP |
| p38 MAPK | p38 mitogen-activated protein kinases |
| PAK/JNK | p21-activated kinase/c-Jun amino terminal kinase |
| Pax7 | Paired box 7 |
| PFN | Profilin |
| PI3K | Phosphatidylinositol 3-kinase |
| RhoA/Rac1 | Ras homolog family member A/Rac Family Small GTPase 1 |
| ROCK | Rho-associated protein kinase |
| SCAR | Suppressor of cyclic AMP repressor |
| Shh | Sonic Hedgehog |
| SIRT1 | Sirtuin 1 |
| Sra1/Cyfip1 | Steroid receptor RNA activator 1/Cytoplasmic FMRP Interacting Protein 1 |
| SRF | Serum Response Factor |
| TGFβ | Transforming growth factor β |
| Tmod | Tropomodulin |
| Tβ4 | Thymosin β4 |
| Toca | Transducer of Cdc42-mediated actin assembly |
| Tpms | Tropomyosins |
| TWF | Twinfilin |
| WASP | Wiskott—Aldrich syndrome protein |
| WAVE | WASP family verprolin homologous protein |
| WDR1 | WD-repeat domain 1 |
| WIP | WASP-interacting protein |
| WRC | WAVE regulatory complex |
| XIN | XIN-repeating protein |
| YAP | Yes-associated protein 1 |
| α-CAA | α-smooth muscles actin |
| α-SKA | α-skeletal muscle actin |
References
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015, 21, 237–248. [Google Scholar] [CrossRef]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Olson, E.N. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA 1996, 93, 9366–9373. [Google Scholar] [CrossRef]
- Yun, K.; Wold, B. Skeletal muscle determination and differentiation: Story of a core regulatory network and its context. Curr. Opin. Cell Biol. 1996, 8, 877–889. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, B.; Ensign, W.Y.; Vogt, P.K.; Han, J. Myogenic differentiation requires signalling through both phosphatidylinositol 3-kinase and p38 MAP kinase. Cell. Signal. 2000, 12, 751–757. [Google Scholar] [CrossRef]
- Wu, Z.; Woodring, P.J.; Bhakta, K.S.; Tamura, K.; Wen, F.; Feramisco, J.R.; Karin, M.; Wang, J.Y.; Puri, P.L. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 2000, 20, 3951–3964. [Google Scholar] [CrossRef]
- Melendez, J.; Sieiro, D.; Salgado, D.; Morin, V.; Dejardin, M.J.; Zhou, C.; Mullen, A.C.; Marcelle, C. TGFbeta signalling acts as a molecular brake of myoblast fusion. Nat. Commun. 2021, 12, 749. [Google Scholar] [CrossRef]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [PubMed]
- von Maltzahn, J.; Chang, N.C.; Bentzinger, C.F.; Rudnicki, M.A. Wnt signaling in myogenesis. Trends Cell Biol. 2012, 22, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Cisternas, P.; Henriquez, J.P.; Brandan, E.; Inestrosa, N.C. Wnt signaling in skeletal muscle dynamics: Myogenesis, neuromuscular synapse and fibrosis. Mol. Neurobiol. 2014, 49, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Rando, T.A. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 2002, 3, 397–409. [Google Scholar] [CrossRef]
- Jang, Y.N.; Baik, E.J. JAK-STAT pathway and myogenic differentiation. JAKSTAT 2013, 2, e23282. [Google Scholar] [CrossRef]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E363–E371. [Google Scholar] [CrossRef]
- Guerin, C.M.; Kramer, S.G. Cytoskeletal remodeling during myotube assembly and guidance: Coordinating the actin and microtubule networks. Commun. Integr. Biol. 2009, 2, 452–457. [Google Scholar] [CrossRef]
- Ono, S. Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton 2010, 67, 677–692. [Google Scholar] [CrossRef]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport. Sci. Rev. 2018, 46, 92–96. [Google Scholar] [CrossRef]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-Mediated Mechanotransduction in Skeletal Muscle. Front. Physiol. 2016, 7, 41. [Google Scholar] [CrossRef]
- Heng, Y.W.; Koh, C.G. Actin cytoskeleton dynamics and the cell division cycle. Int. J. Biochem. Cell Biol. 2010, 42, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Nakamura, F. Actin-Associated Proteins and Small Molecules Targeting the Actin Cytoskeleton. Int. J. Mol. Sci. 2022, 23, 2118. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [PubMed]
- dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef]
- Mohri, K.; Takano-Ohmuro, H.; Nakashima, H.; Hayakawa, K.; Endo, T.; Hanaoka, K.; Obinata, T. Expression of cofilin isoforms during development of mouse striated muscles. J. Muscle Res. Cell Motil. 2000, 21, 49–57. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, H.; Zhao, S.; Zhang, J.; Liu, D.; Tian, Y.; Shen, Z.; Su, Y. Role of the cofilin 2 gene in regulating the myosin heavy chain genes in mouse myoblast C2C12 cells. Int. J. Mol. Med. 2018, 41, 1096–1102. [Google Scholar] [CrossRef]
- Kepser, L.J.; Damar, F.; De Cicco, T.; Chaponnier, C.; Proszynski, T.J.; Pagenstecher, A.; Rust, M.B. CAP2 deficiency delays myofibril actin cytoskeleton differentiation and disturbs skeletal muscle architecture and function. Proc. Natl. Acad. Sci. USA 2019, 116, 8397–8402. [Google Scholar] [CrossRef]
- Nowak, S.J.; Nahirney, P.C.; Hadjantonakis, A.K.; Baylies, M.K. Nap1-mediated actin remodeling is essential for mammalian myoblast fusion. J. Cell Sci. 2009, 122, 3282–3293. [Google Scholar] [CrossRef]
- Kawamura, K.; Takano, K.; Suetsugu, S.; Kurisu, S.; Yamazaki, D.; Miki, H.; Takenawa, T.; Endo, T. N-WASP and WAVE2 acting downstream of phosphatidylinositol 3-kinase are required for myogenic cell migration induced by hepatocyte growth factor. J. Biol. Chem. 2004, 279, 54862–54871. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Won, Y.H.; Kwon, T.W.; Lee, W. Twinfilin-1 is an essential regulator of myogenic differentiation through the modulation of YAP in C2C12 myoblasts. Biochem. Biophys. Res. Commun. 2022, 599, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef]
- Ojima, K.; Lin, Z.X.; Andrade, I.R.; Costa, M.L.; Mermelstein, C. Distinctive Effects of Cytochalasin B in Chick Primary Myoblasts and Fibroblasts. PLoS ONE 2016, 11, e0154109. [Google Scholar] [CrossRef]
- Sanger, J.W. The use of cytochalasin B to distinguish myoblasts from fibroblasts in cultures of developing chick striated muscle. Proc. Natl. Acad. Sci. USA 1974, 71, 3621–3625. [Google Scholar] [CrossRef]
- Wang, J.; Sanger, J.M.; Sanger, J.W. Differential effects of Latrunculin-A on myofibrils in cultures of skeletal muscle cells: Insights into mechanisms of myofibrillogenesis. Cell Motil. Cytoskel 2005, 62, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, Y.; Dube, D.K.; Sanger, J.M.; Sanger, J.W. Jasplakinolide reduces actin and tropomyosin dynamics during myofibrillogenesis. Cytoskeleton 2014, 71, 513–529. [Google Scholar] [CrossRef]
- Witke, W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004, 14, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.; Nyström, L.-E.; Sundkvist, I.; Markey, F.; Lindberg, U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J. Mol. Biol. 1977, 115, 465–483. [Google Scholar] [CrossRef]
- Lappalainen, P.; Kessels, M.M.; Cope, M.J.; Drubin, D.G. The ADF homology (ADF-H) domain: A highly exploited actin-binding module. Mol. Biol. Cell 1998, 9, 1951–1959. [Google Scholar] [CrossRef]
- Bamburg, J.R. Proteins of the ADF/cofilin family: Essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999, 15, 185–230. [Google Scholar] [CrossRef]
- Ono, S. Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: New blades for twisted filaments. Biochemistry 2003, 42, 13363–13370. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Blanchoin, L.; Abe, H.; Chen, H.; Pollard, T.D.; Bamburg, J.R. Xenopus actin-interacting protein 1 (XAip1) enhances cofilin fragmentation of filaments by capping filament ends. J. Biol. Chem. 2002, 277, 43011–43016. [Google Scholar] [CrossRef]
- Goode, B.L.; Drubin, D.G.; Lappalainen, P. Regulation of the cortical actin cytoskeleton in budding yeast by twinfilin, a ubiquitous actin monomer-sequestering protein. J. Cell Biol. 1998, 142, 723–733. [Google Scholar] [CrossRef]
- Vartiainen, M.; Ojala, P.J.; Auvinen, P.; Peranen, J.; Lappalainen, P. Mouse A6/twinfilin is an actin monomer-binding protein that localizes to the regions of rapid actin dynamics. Mol. Cell. Biol. 2000, 20, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Helfer, E.; Nevalainen, E.M.; Naumanen, P.; Romero, S.; Didry, D.; Pantaloni, D.; Lappalainen, P.; Carlier, M.F. Mammalian twinfilin sequesters ADP-G-actin and caps filament barbed ends: Implications in motility. EMBO J. 2006, 25, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Peche, V.; Shekar, S.; Leichter, M.; Korte, H.; Schroder, R.; Schleicher, M.; Holak, T.A.; Clemen, C.S.; Ramanath, Y.B.; Pfitzer, G.; et al. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell Mol. Life Sci. 2007, 64, 2702–2715. [Google Scholar] [CrossRef]
- Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics—More than a monomer-sequestration factor. J. Cell Sci. 2013, 126, 3249–3258. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Zigmond, S.H.; Evangelista, M.; Boone, C.; Yang, C.; Dar, A.C.; Sicheri, F.; Forkey, J.; Pring, M. Formin leaky cap allows elongation in the presence of tight capping proteins. Curr. Biol. 2003, 13, 1820–1823. [Google Scholar] [CrossRef]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef]
- Mullins, R.D.; Pollard, T.D. Structure and function of the Arp2/3 complex. Curr. Opin. Struct. Biol. 1999, 9, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Daugherty-Clarke, K.; Goode, B.L.; Gelles, J. Pathway of actin filament branch formation by Arp2/3 complex revealed by single-molecule imaging. Proc. Natl. Acad. Sci. USA 2013, 110, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Insall, R.H. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 1998, 8, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Rodal, A.A.; Barnes, G.; Drubin, D.G. Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J. Cell Biol. 2001, 153, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.A.; Korshunova, Y.O.; Schroer, T.A.; Cooper, J.A. Differential localization and sequence analysis of capping protein beta-subunit isoforms of vertebrates. J. Cell Biol. 1994, 127, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Papa, I.; Astier, C.; Kwiatek, O.; Raynaud, F.; Bonnal, C.; Lebart, M.C.; Roustan, C.; Benyamin, Y. Alpha actinin-CapZ, an anchoring complex for thin filaments in Z-line. J. Muscle Res. Cell Motil. 1999, 20, 187–197. [Google Scholar] [CrossRef]
- McGough, A.M.; Staiger, C.J.; Min, J.K.; Simonetti, K.D. The gelsolin family of actin regulatory proteins: Modular structures, versatile functions. FEBS Lett. 2003, 552, 75–81. [Google Scholar] [CrossRef]
- Webert, A.; Pennise, C.; Fowler, V. Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting adp. Pi-actin to adp-actin at all pointed filament ends. J. Biol. Chem. 1999, 274, 34637–34645. [Google Scholar] [CrossRef]
- Yamashiro, S.; Yamakita, Y.; Ono, S.; Matsumura, F. Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Mol. Biol. Cell 1998, 9, 993–1006. [Google Scholar] [CrossRef]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef]
- Daimon, E.; Shibukawa, Y.; Wada, Y. Calponin 3 regulates stress fiber formation in dermal fibroblasts during wound healing. Arch. Dermatol. Res. 2013, 305, 571–584. [Google Scholar] [CrossRef]
- Majoul, I.; Shirao, T.; Sekino, Y.; Duden, R. Many faces of drebrin: From building dendritic spines and stabilizing gap junctions to shaping neurite-like cell processes. Histochem. Cell Biol. 2007, 127, 355–361. [Google Scholar] [CrossRef]
- Giridharan, S.S.; Caplan, S. MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid. Redox Signal. 2014, 20, 2059–2073. [Google Scholar] [CrossRef]
- Terman, J.R.; Mao, T.; Pasterkamp, R.J.; Yu, H.H.; Kolodkin, A.L. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 2002, 109, 887–900. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Nakanishi, H.; Ikeda, W.; Satoh, A.; Momose, Y.; Nishioka, H.; Takai, Y. Nexilin: A novel actin filament-binding protein localized at cell-matrix adherens junction. J. Cell Biol. 1998, 143, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Pacholsky, D.; Vakeel, P.; Himmel, M.; Lowe, T.; Stradal, T.; Rottner, K.; Furst, D.O.; van der Ven, P.F. Xin repeats define a novel actin-binding motif. J. Cell Sci. 2004, 117, 5257–5268. [Google Scholar] [CrossRef] [PubMed]
- Gokhin, D.S.; Fowler, V.M. Tropomodulin capping of actin filaments in striated muscle development and physiology. J. Biomed. Biotechnol. 2011, 2011, 103069. [Google Scholar] [CrossRef]
- Fowler, V.M.; Dominguez, R. Tropomodulins and Leiomodins: Actin Pointed End Caps and Nucleators in Muscles. Biophys. J. 2017, 112, 1742–1760. [Google Scholar] [CrossRef]
- Li, F.; Kolb, J.; Crudele, J.; Tonino, P.; Hourani, Z.; Smith, J.E., 3rd; Chamberlain, J.S.; Granzier, H. Expressing a Z-disk nebulin fragment in nebulin-deficient mouse muscle: Effects on muscle structure and function. Skelet. Muscle 2020, 10, 2. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Granzier, H. New Insights into the Structural Roles of Nebulin in Skeletal Muscle. J. Biomed. Biotechnol. 2010, 2010, 1–6. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, A.S.; Kazmierski, S.T.; Labeit, S.; Gregorio, C.C. Nebulin: The nebulous, multifunctional giant of striated muscle. Trends Cardiovasc. Med. 2003, 13, 195–201. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Granzier, H.; Labeit, S. The sarcomeric protein nebulin: Another multifunctional giant in charge of muscle strength optimization. Front. Physiol. 2012, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Choi, Y.N.; Seo, T.W.; Lee, J.S.; Yoo, S.J. Ubiquitin-proteasome dependent regulation of Profilin2 (Pfn2) by a cellular inhibitor of apoptotic protein 1 (cIAP1). Biochem. Biophys. Res. Commun. 2018, 506, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Witke, W.; Podtelejnikov, A.V.; Di Nardo, A.; Sutherland, J.D.; Gurniak, C.B.; Dotti, C.; Mann, M. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 1998, 17, 967–976. [Google Scholar] [CrossRef]
- Lambrechts, A.; Braun, A.; Jonckheere, V.; Aszodi, A.; Lanier, L.M.; Robbens, J.; Van Colen, I.; Vandekerckhove, J.; Fassler, R.; Ampe, C. Profilin II is alternatively spliced, resulting in profilin isoforms that are differentially expressed and have distinct biochemical properties. Mol. Cell Biol. 2000, 20, 8209–8219. [Google Scholar] [CrossRef]
- Ayscough, K.R. In vivo functions of actin-binding proteins. Curr. Opin. Cell Biol. 1998, 10, 102–111. [Google Scholar] [CrossRef]
- Jasper, H.; Benes, V.; Schwager, C.; Sauer, S.; Clauder-Munster, S.; Ansorge, W.; Bohmann, D. The genomic response of the Drosophila embryo to JNK signaling. Dev. Cell 2001, 1, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.; Rubenstein, P.A. Control of profilin and actin expression in muscle and nonmuscle cells. Cell Motil. Cytoskel 1993, 24, 179–188. [Google Scholar] [CrossRef]
- Gopinath, S.D.; Narumiya, S.; Dhawan, J. The RhoA effector mDiaphanous regulates MyoD expression and cell cycle progression via SRF-dependent and SRF-independent pathways. J. Cell Sci. 2007, 120, 3086–3098. [Google Scholar] [CrossRef]
- L’Honore, A.; Rana, V.; Arsic, N.; Franckhauser, C.; Lamb, N.J.; Fernandez, A. Identification of a new hybrid serum response factor and myocyte enhancer factor 2-binding element in MyoD enhancer required for MyoD expression during myogenesis. Mol. Biol. Cell 2007, 18, 1992–2001. [Google Scholar] [CrossRef]
- Zi, J.; Xu, J.; Luo, J.; Yang, X.; Zhen, Z.; Li, X.; Hu, D.; Guo, Y.; Guo, H.; Ding, X.; et al. PFN1 Inhibits Myogenesis of Bovine Myoblast Cells via Cdc42-PAK/JNK. Cells 2022, 11, 3188. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Viswanathan, M.C.; Lee, D.I.; Rainer, P.P.; Schmidt, W.; Kronert, W.A.; Harding, S.E.; Kass, D.A.; Bernstein, S.I.; Van Eyk, J.E.; et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 110, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hou, L.; Zhang, Y.; Jiang, F.; Zhu, Y.; Li, Q.X.; Hu, C.Y.; Wang, C. PFN2a Suppresses C2C12 Myogenic Development by Inhibiting Proliferation and Promoting Apoptosis via the p53 Pathway. Cells 2019, 8, 959. [Google Scholar] [CrossRef] [PubMed]
- McGough, A.; Pope, B.; Chiu, W.; Weeds, A. Cofilin changes the twist of F-actin: Implications for actin filament dynamics and cellular function. J. Cell Biol. 1997, 138, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Maciver, S.K.; Hussey, P.J. The ADF/cofilin family: Actin-remodeling proteins. Genome Biol. 2002, 3, reviews3007. [Google Scholar] [CrossRef]
- Maggi, L.; Scoto, M.; Cirak, S.; Robb, S.A.; Klein, A.; Lillis, S.; Cullup, T.; Feng, L.; Manzur, A.Y.; Sewry, C.A.; et al. Congenital myopathies--clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul. Disord. 2013, 23, 195–205. [Google Scholar] [CrossRef]
- Malfatti, E.; Romero, N.B. Nemaline myopathies: State of the art. Rev. Neurol. 2016, 172, 614–619. [Google Scholar] [CrossRef]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, Y.; Zhao, T.; Li, M.; Mao, Y.; Yang, Z. Epigenetic Regulation Mechanisms of the Cofilin-1 Gene in the Development and Differentiation of Bovine Primary Myoblasts. Genes 2022, 13, 723. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Min, K.H.; Kim, D.; Park, S.Y.; Lee, W. CFL2 is an essential mediator for myogenic differentiation in C2C12 myoblasts. Biochem. Biophys. Res. Commun. 2020, 533, 710–716. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Yu, S.F.; Chin, S.M.; Soffar, D.B.; Windner, S.E.; Goode, B.L.; Baylies, M.K. Cofilin Loss in Drosophila Muscles Contributes to Muscle Weakness through Defective Sarcomerogenesis during Muscle Growth. Cell Rep. 2020, 32, 107893. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Joshi, M.; Savic, T.; Beggs, A.H.; Agrawal, P.B. Skeletal muscle microRNA and messenger RNA profiling in cofilin-2 deficient mice reveals cell cycle dysregulation hindering muscle regeneration. PLoS ONE 2015, 10, e0123829. [Google Scholar] [CrossRef] [PubMed]
- Gurniak, C.B.; Chevessier, F.; Jokwitz, M.; Jonsson, F.; Perlas, E.; Richter, H.; Matern, G.; Boyl, P.P.; Chaponnier, C.; Furst, D.; et al. Severe protein aggregate myopathy in a knockout mouse model points to an essential role of cofilin2 in sarcomeric actin exchange and muscle maintenance. Eur. J. Cell Biol. 2014, 93, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.B.; Joshi, M.; Savic, T.; Chen, Z.; Beggs, A.H. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 2012, 21, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Kremneva, E.; Makkonen, M.H.; Skwarek-Maruszewska, A.; Gateva, G.; Michelot, A.; Dominguez, R.; Lappalainen, P. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev. Cell 2014, 31, 215–226. [Google Scholar] [CrossRef]
- de Winter, J.M.; Ottenheijm, C.A.C. Sarcomere Dysfunction in Nemaline Myopathy. J. Neuromuscul. Dis. 2017, 4, 99–113. [Google Scholar] [CrossRef]
- Salanova, M.; Gelfi, C.; Moriggi, M.; Vasso, M.; Vigano, A.; Minafra, L.; Bonifacio, G.; Schiffl, G.; Gutsmann, M.; Felsenberg, D.; et al. Disuse deterioration of human skeletal muscle challenged by resistive exercise superimposed with vibration: Evidence from structural and proteomic analysis. FASEB J. 2014, 28, 4748–4763. [Google Scholar] [CrossRef]
- Moriyama, K.; Yahara, I. Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell Sci. 2002, 115, 1591–1601. [Google Scholar] [CrossRef]
- Bertling, E.; Hotulainen, P.; Mattila, P.K.; Matilainen, T.; Salminen, M.; Lappalainen, P. Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 2004, 15, 2324–2334. [Google Scholar] [CrossRef]
- Singh, A.K.; Rai, A.; Weber, A.; Posern, G. miRNA mediated downregulation of cyclase-associated protein 1 (CAP1) is required for myoblast fusion. Front. Cell Dev. Biol. 2022, 10, 899917. [Google Scholar] [CrossRef]
- Sun, Y.N.; Huang, J.Q.; Chen, Z.Z.; Du, M.; Ren, F.Z.; Luo, J.; Fang, B. Amyotrophy Induced by a High-Fat Diet Is Closely Related to Inflammation and Protein Degradation Determined by Quantitative Phosphoproteomic Analysis in Skeletal Muscle of C57BL/6 J Mice. J. Nutr. 2020, 150, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, E.M.; Skwarek-Maruszewska, A.; Braun, A.; Moser, M.; Lappalainen, P. Two biochemically distinct and tissue-specific twinfilin isoforms are generated from the mouse Twf2 gene by alternative promoter usage. Biochem. J. 2009, 417, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Vartiainen, M.K.; Sarkkinen, E.M.; Matilainen, T.; Salminen, M.; Lappalainen, P. Mammals have two twinfilin isoforms whose subcellular localizations and tissue distributions are differentially regulated. J. Biol. Chem. 2003, 278, 34347–34355. [Google Scholar] [CrossRef] [PubMed]
- Safer, D.; Elzinga, M.; Nachmias, V.T. Thymosin beta 4 and Fx, an actin-sequestering peptide, are indistinguishable. J. Biol. Chem. 1991, 266, 4029–4032. [Google Scholar] [CrossRef]
- Malinda, K.M.; Sidhu, G.S.; Mani, H.; Banaudha, K.; Maheshwari, R.K.; Goldstein, A.L.; Kleinman, H.K. Thymosin beta4 accelerates wound healing. J. Investig. Dermatol. 1999, 113, 364–368. [Google Scholar] [CrossRef]
- Philp, D.; Nguyen, M.; Scheremeta, B.; St-Surin, S.; Villa, A.M.; Orgel, A.; Kleinman, H.K.; Elkin, M. Thymosin beta4 increases hair growth by activation of hair follicle stem cells. FASEB J. 2004, 18, 385–387. [Google Scholar] [CrossRef]
- Bock-Marquette, I.; Saxena, A.; White, M.D.; Dimaio, J.M.; Srivastava, D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 2004, 432, 466–472. [Google Scholar] [CrossRef]
- Tseng, B.S.; Zhao, P.; Pattison, J.S.; Gordon, S.E.; Granchelli, J.A.; Madsen, R.W.; Folk, L.C.; Hoffman, E.P.; Booth, F.W. Regenerated mdx mouse skeletal muscle shows differential mRNA expression. J. Appl. Physiol. 2002, 93, 537–545. [Google Scholar] [CrossRef]
- Turk, R.; Sterrenburg, E.; de Meijer, E.J.; van Ommen, G.J.; den Dunnen, J.T.; t Hoen, P.A. Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC Genom. 2005, 6, 98. [Google Scholar] [CrossRef]
- Nakayama, Y.; Nara, N.; Kawakita, Y.; Takeshima, Y.; Arakawa, M.; Katoh, M.; Morita, S.; Iwatsuki, K.; Tanaka, K.; Okamoto, S.; et al. Cloning of cDNA encoding a regeneration-associated muscle protease whose expression is attenuated in cell lines derived from Duchenne muscular dystrophy patients. Am. J. Pathol. 2004, 164, 1773–1782. [Google Scholar] [CrossRef]
- Tokura, Y.; Nakayama, Y.; Fukada, S.-i.; Nara, N.; Yamamoto, H.; Matsuda, R.; Hara, T. Muscle injury-induced thymosin β4 acts as a chemoattractant for myoblasts. J. Biochem. 2011, 149, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Franquesa, A.; Stocks, B.; Borg, M.L.; Kuefner, M.; Dalbram, E.; Nielsen, T.S.; Agrawal, A.; Pankratova, S.; Chibalin, A.V.; Karlsson, H.K. Discovery of thymosin β4 as a human exerkine and growth factor. Am. J. Physiol.-Cell Physiol. 2021, 321, C770–C778. [Google Scholar] [CrossRef]
- Sun, H.Q.; Kwiatkowska, K.; Yin, H.L. beta-Thymosins are not simple actin monomer buffering proteins. Insights from overexpression studies. J. Biol. Chem. 1996, 271, 9223–9230. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Kuzumaki, N. Transcriptional activity of megakaryoblastic leukemia 1 (MKL1) is repressed by SUMO modification. Genes. Cells 2005, 10, 835–850. [Google Scholar] [CrossRef]
- Muehlich, S.; Wang, R.; Lee, S.M.; Lewis, T.C.; Dai, C.; Prywes, R. Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol. Cell Biol. 2008, 28, 6302–6313. [Google Scholar] [CrossRef]
- Charbonney, E.; Speight, P.; Masszi, A.; Nakano, H.; Kapus, A. beta-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol. Biol. Cell 2011, 22, 4472–4485. [Google Scholar] [CrossRef]
- Cenik, B.K.; Liu, N.; Chen, B.; Bezprozvannaya, S.; Olson, E.N.; Bassel-Duby, R. Myocardin-related transcription factors are required for skeletal muscle development. Development 2016, 143, 2853–2861. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.; Prywes, R. Megakaryoblastic leukemia-1/2, a transcriptional co-activator of serum response factor, is required for skeletal myogenic differentiation. J. Biol. Chem. 2003, 278, 41977–41987. [Google Scholar] [CrossRef]
- Iwasaki, K.; Hayashi, K.; Fujioka, T.; Sobue, K. Rho/Rho-associated kinase signal regulates myogenic differentiation via myocardin-related transcription factor-A/Smad-dependent transcription of the Id3 gene. J. Biol. Chem. 2008, 283, 21230–21241. [Google Scholar] [CrossRef]
- Li, S.; Czubryt, M.P.; McAnally, J.; Bassel-Duby, R.; Richardson, J.A.; Wiebel, F.F.; Nordheim, A.; Olson, E.N. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1082–1087. [Google Scholar] [CrossRef]
- Mokalled, M.H.; Johnson, A.N.; Creemers, E.E.; Olson, E.N. MASTR directs MyoD-dependent satellite cell differentiation during skeletal muscle regeneration. Genes. Dev. 2012, 26, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, K.; Akiho, M.; Nakashima, H.; Akima, H.; Yasuhara, M. Age-related reductions in expression of serum response factor and myocardin-related transcription factor A in mouse skeletal muscles. Biochim. Biophys. Acta 2008, 1782, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Barrientos, T.; Pipes, G.C.; Li, S.; Olson, E.N. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol. Cell Biol. 2005, 25, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Descot, A.; Hoffmann, R.; Shaposhnikov, D.; Reschke, M.; Ullrich, A.; Posern, G. Negative regulation of the EGFR-MAPK cascade by actin-MAL-mediated Mig6/Errfi-1 induction. Mol. Cell 2009, 35, 291–304. [Google Scholar] [CrossRef]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes. Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Charrasse, S.; Comunale, F.; Grumbach, Y.; Poulat, F.; Blangy, A.; Gauthier-Rouviere, C. RhoA GTPase regulates M-cadherin activity and myoblast fusion. Mol. Biol. Cell 2006, 17, 749–759. [Google Scholar] [CrossRef]
- Song, R.; Zhao, S.; Xu, Y.; Hu, J.; Ke, S.; Li, F.; Tian, G.; Zheng, X.; Li, J.; Gu, L.; et al. MRTF-A regulates myoblast commitment to differentiation by targeting PAX7 during muscle regeneration. J. Cell Mol. Med. 2021, 25, 8645–8661. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, P.; Duan, R.; Chen, E.H. Mechanisms of myoblast fusion during muscle development. Curr. Opin. Genet. Dev. 2015, 32, 162–170. [Google Scholar] [CrossRef]
- Richardson, B.E.; Beckett, K.; Nowak, S.J.; Baylies, M.K. SCAR/WAVE and Arp2/3 are crucial for cytoskeletal remodeling at the site of myoblast fusion. Development 2007, 134, 4357–4367. [Google Scholar] [CrossRef]
- Stradal, T.E.; Scita, G. Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell Biol. 2006, 18, 4–10. [Google Scholar] [CrossRef]
- Bompard, G.; Caron, E. Regulation of WASP/WAVE proteins: Making a long story short. J. Cell Biol. 2004, 166, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Miki, H. WASP and WAVE family proteins: Key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Benesch, S.; Lommel, S.; Steffen, A.; Stradal, T.E.; Scaplehorn, N.; Way, M.; Wehland, J.; Rottner, K. Phosphatidylinositol 4,5-biphosphate (PIP2)-induced vesicle movement depends on N-WASP and involves Nck, WIP, and Grb2. J. Biol. Chem. 2002, 277, 37771–37776. [Google Scholar] [CrossRef]
- Miki, H.; Yamaguchi, H.; Suetsugu, S.; Takenawa, T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 2000, 408, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and control of the actin regulatory WAVE complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Gruenbaum-Cohen, Y.; Harel, I.; Umansky, K.B.; Tzahor, E.; Snapper, S.B.; Shilo, B.Z.; Schejter, E.D. The actin regulator N-WASp is required for muscle-cell fusion in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 11211–11216. [Google Scholar] [CrossRef]
- Mitra, P.; Thanabalu, T. Myogenic differentiation depends on the interplay of Grb2 and N-WASP. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 487–497. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Jain, N.; Fen Chong, P.; Hou Tan, J.; Thanabalu, T. Myogenesis defect due to Toca-1 knockdown can be suppressed by expression of N-WASP. Biochim. Biophys. Acta 2014, 1843, 1930–1941. [Google Scholar] [CrossRef]
- Bacon, C.; Lakics, V.; Machesky, L.; Rumsby, M. N-WASP regulates extension of filopodia and processes by oligodendrocyte progenitors, oligodendrocytes, and Schwann cells-implications for axon ensheathment at myelination. Glia 2007, 55, 844–858. [Google Scholar] [CrossRef]
- Miki, H.; Sasaki, T.; Takai, Y.; Takenawa, T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 1998, 391, 93–96. [Google Scholar] [CrossRef]
- Snapper, S.B.; Takeshima, F.; Anton, I.; Liu, C.H.; Thomas, S.M.; Nguyen, D.; Dudley, D.; Fraser, H.; Purich, D.; Lopez-Ilasaca, M.; et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 2001, 3, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Vasyutina, E.; Martarelli, B.; Brakebusch, C.; Wende, H.; Birchmeier, C. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 8935–8940. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.F.; Nioche, P.; Broutin-L’Hermite, I.; Boujemaa, R.; Le Clainche, C.; Egile, C.; Garbay, C.; Ducruix, A.; Sansonetti, P.; Pantaloni, D. GRB2 links signaling to actin assembly by enhancing interaction of neural Wiskott-Aldrich syndrome protein (N-WASp) with actin-related protein (ARP2/3) complex. J. Biol. Chem. 2000, 275, 21946–21952. [Google Scholar] [CrossRef] [PubMed]
- Leshem, Y.; Gitelman, I.; Ponzetto, C.; Halevy, O. Preferential binding of Grb2 or phosphatidylinositol 3-kinase to the met receptor has opposite effects on HGF-induced myoblast proliferation. Exp. Cell Res. 2002, 274, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Spicer, D.B.; Rhee, J.; Cheung, W.L.; Lassar, A.B. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science 1996, 272, 1476–1480. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Leroy, M.C.; Perroud, J.; Darbellay, B.; Bernheim, L.; Konig, S. Epidermal growth factor receptor down-regulation triggers human myoblast differentiation. PLoS ONE 2013, 8, e71770. [Google Scholar] [CrossRef]
- Ho, H.Y.; Rohatgi, R.; Lebensohn, A.M.; Le, M.; Li, J.; Gygi, S.P.; Kirschner, M.W. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell 2004, 118, 203–216. [Google Scholar] [CrossRef]
- Vavylonis, D.; Kovar, D.R.; O’Shaughnessy, B.; Pollard, T.D. Model of formin-associated actin filament elongation. Mol. Cell 2006, 21, 455–466. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical roles for multiple formins during cardiac myofibril development and repair. Mol. Biol. Cell 2014, 25, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Spletter, M.L.; Barz, C.; Yeroslaviz, A.; Zhang, X.; Lemke, S.B.; Bonnard, A.; Brunner, E.; Cardone, G.; Basler, K.; Habermann, B.H.; et al. A transcriptomics resource reveals a transcriptional transition during ordered sarcomere morphogenesis in flight muscle. Elife 2018, 7, e34058. [Google Scholar] [CrossRef] [PubMed]
- Al Haj, A.; Mazur, A.J.; Radaszkiewicz, K.; Radaszkiewicz, T.; Makowiecka, A.; Stopschinski, B.E.; Schonichen, A.; Geyer, M.; Mannherz, H.G. Distribution of formins in cardiac muscle: FHOD1 is a component of intercalated discs and costameres. Eur. J. Cell Biol. 2015, 94, 101–113. [Google Scholar] [CrossRef]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan, O.M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef] [PubMed]
- Shwartz, A.; Dhanyasi, N.; Schejter, E.D.; Shilo, B.Z. The Drosophila formin Fhos is a primary mediator of sarcomeric thin-filament array assembly. Elife 2016, 5, e16540. [Google Scholar] [CrossRef]
- Kan-o, M.; Takeya, R.; Taniguchi, K.; Tanoue, Y.; Tominaga, R.; Sumimoto, H. Expression and subcellular localization of mammalian formin Fhod3 in the embryonic and adult heart. PLoS ONE 2012, 7, e34765. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; Garcia-Pinilla, J.M.; Mogensen, J.; Restrepo-Cordoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef]
- Moseley, J.B.; Sagot, I.; Manning, A.L.; Xu, Y.W.; Eck, J.; Pellman, D.; Goode, B.L. A conserved mechanism for Bni1-and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell 2004, 15, 896–907. [Google Scholar] [CrossRef]
- Vig, A.T.; Foldi, I.; Szikora, S.; Migh, E.; Gombos, R.; Toth, M.A.; Huber, T.; Pinter, R.; Talian, G.C.; Mihaly, J.; et al. The activities of the C-terminal regions of the formin protein disheveled-associated activator of morphogenesis (DAAM) in actin dynamics. J. Biol. Chem. 2017, 292, 13566–13583. [Google Scholar] [CrossRef]
- Molnar, I.; Migh, E.; Szikora, S.; Kalmar, T.; Vegh, A.G.; Deak, F.; Barko, S.; Bugyi, B.; Orfanos, Z.; Kovacs, J.; et al. DAAM Is Required for Thin Filament Formation and Sarcomerogenesis during Muscle Development in Drosophila. PLoS Genet. 2014, 10, e1004166. [Google Scholar] [CrossRef]
- Deng, S.; Silimon, R.L.; Balakrishnan, M.; Bothe, I.; Juros, D.; Soffar, D.B.; Baylies, M.K. The actin polymerization factor Diaphanous and the actin severing protein Flightless I collaborate to regulate sarcomere size. Dev. Biol. 2021, 469, 12–25. [Google Scholar] [CrossRef]
- Deng, S.; Bothe, I.; Baylies, M.K. The Formin Diaphanous Regulates Myoblast Fusion through Actin Polymerization and Arp2/3 Regulation. PLoS Genet. 2015, 11, e1005381. [Google Scholar] [CrossRef]
- Szikora, S.; Gorog, P.; Mihaly, J. The Mechanisms of Thin Filament Assembly and Length Regulation in Muscles. Int. J. Mol. Sci. 2022, 23, 5306. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, S.; Tomasevic, N.; Weed, S.; Sakowicz, R.; Cooper, J.A. Src phosphorylation of cortactin enhances actin assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 11933–11938. [Google Scholar] [CrossRef]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.; Egile, C.; Li, R.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ma, W.; An, L. Cortactin in cancer cell migration and invasion. Oncotarget 2017, 8, 88232–88243. [Google Scholar] [CrossRef] [PubMed]
- Nazari, H.; Khaleghian, A.; Takahashi, A.; Harada, N.; Webster, N.J.; Nakano, M.; Kishi, K.; Ebina, Y.; Nakaya, Y. Cortactin, an actin binding protein, regulates GLUT4 translocation via actin filament remodeling. Biochemistry 2011, 76, 1262–1269. [Google Scholar] [CrossRef]
- Iwahara, N.; Azekami, K.; Hosoda, R.; Nojima, I.; Hisahara, S.; Kuno, A. Activation of SIRT1 promotes membrane resealing via cortactin. Sci. Rep. 2022, 12, 15328. [Google Scholar] [CrossRef]
- Bisht, B.; Dey, C.S. Focal Adhesion Kinase contributes to insulin-induced actin reorganization into a mesh harboring Glucose transporter-4 in insulin resistant skeletal muscle cells. Bmc Cell Biol. 2008, 9, 48. [Google Scholar] [CrossRef]
- Ai, H.; Ralston, E.; Lauritzen, H.P.; Galbo, H.; Ploug, T. Disruption of microtubules in rat skeletal muscle does not inhibit insulin- or contraction-stimulated glucose transport. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E836–E844. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, D.; Iwahara, N.; Sebori, R.; Hosoda, R.; Shimohama, S.; Kuno, A.; Horio, Y. SIRT1 deficiency interferes with membrane resealing after cell membrane injury. PLoS ONE 2019, 14, e0218329. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, R.; Jimbow, K.; Tanimura, A.; Sato, M.; Horimoto, K.; Hayashi, T.; Hisahara, S.; Sugino, T.; Hirobe, T.; Yamashita, T.; et al. SIRT1 regulates lamellipodium extension and migration of melanoma cells. J. Investig. Dermatol. 2014, 134, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.; Mehl, R.; Izumo, S.; Nadal-Ginard, B.; Yin, H. Muscle is the major source of plasma gelsolin. J. Biol. Chem. 1988, 263, 8239–8243. [Google Scholar] [CrossRef]
- Bertin, B.; Renaud, Y.; Jagla, T.; Lavergne, G.; Dondi, C.; Da Ponte, J.P.; Junion, G.; Jagla, K. Gelsolin and dCryAB act downstream of muscle identity genes and contribute to preventing muscle splitting and branching in Drosophila. Sci. Rep. 2021, 11, 13197. [Google Scholar] [CrossRef]
- Scholz, A.; Hinssen, H. Biphasic pattern of gelsolin expression and variations in gelsolin-actin interactions during myogenesis. Exp. Cell Res. 1995, 219, 384–391. [Google Scholar] [CrossRef]
- Hishiya, A.; Kitazawa, T.; Takayama, S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ. Res. 2010, 107, 1220–1231. [Google Scholar] [CrossRef]
- Fowler, V.M.; Sussmann, M.A.; Miller, P.G.; Flucher, B.E.; Daniels, M.P. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J. Cell Biol. 1993, 120, 411–420. [Google Scholar] [CrossRef]
- Almenar-Queralt, A.; Lee, A.; Conley, C.A.; de Pouplana, L.D.; Fowler, V.M. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J. Biol. Chem. 2000, 275, 13164. [Google Scholar] [CrossRef]
- Gokhin, D.S.; Lewis, R.A.; McKeown, C.R.; Nowak, R.B.; Kim, N.E.; Littlefield, R.S.; Lieber, R.L.; Fowler, V.M. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J. Cell Biol. 2010, 189, 95–109. [Google Scholar] [CrossRef]
- Cox, P.R.; Zoghbi, H.Y. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics 2000, 63, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, Z.; Liu, X.; Chen, Y.; Gong, W.; Yu, K.; Qin, L.; Chen, H.; Mo, D. The switch role of the Tmod4 in the regulation of balanced development between myogenesis and adipogenesis. Gene 2013, 532, 263–271. [Google Scholar] [CrossRef]
- Littlefield, R.; Almenar-Queralt, A.A.; Fowler, V.M. Actin dynamics at pointed ends regulate thin filament length in striated muscle. Mol. Biol. Cell 2001, 3, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Mardahl-Dumesnil, M.; Fowler, V.M. Thin filaments elongate from their pointed ends during myofibril assembly in Drosophila indirect flight muscle. J. Cell Biol. 2001, 155, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M.A.; Welch, S.; Cambon, N.; Klevitsky, R.; Hewett, T.E.; Price, R.; Witt, S.A.; Kimball, T.R. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J. Clin. Investig. 1998, 101, 51–61. [Google Scholar] [CrossRef]
- Gokhin, D.S.; Fowler, V.M. Cytoplasmic gamma-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. J. Cell Biol. 2011, 194, 105–120. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef]
- Yuen, M.; Sandaradura, S.A.; Dowling, J.J.; Kostyukova, A.S.; Moroz, N.; Quinlan, K.G.; Lehtokari, V.L.; Ravenscroft, G.; Todd, E.J.; Ceyhan-Birsoy, O.; et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Investig. 2015, 125, 456–457. [Google Scholar] [CrossRef]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef]
- Lin, F.H.; Wang, A.; Dai, W.; Chen, S.; Ding, Y.; Sun, L.V. Lmod3 promotes myoblast differentiation and proliferation via the AKT and ERK pathways. Exp. Cell Res. 2020, 396, 112297. [Google Scholar] [CrossRef]
- Cenik, B.K.; Garg, A.; McAnally, J.R.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N.; Liu, N. Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Investig. 2015, 125, 1569–1578. [Google Scholar] [CrossRef]
- Small, E.M.; O’Rourke, J.R.; Moresi, V.; Sutherland, L.B.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 2010, 107, 4218–4223. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.H.; Stossel, T.P. Isolation and properties of actin, myosin, and a new actinbinding protein in rabbit alveolar macrophages. J. Biol. Chem. 1975, 250, 5696–5705. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, I.; Kunkel, L.M. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Bonnemann, C.G.; Thompson, T.G.; van der Ven, P.F.; Goebel, H.H.; Warlo, I.; Vollmers, B.; Reimann, J.; Herms, J.; Gautel, M.; Takada, F.; et al. Filamin C accumulation is a strong but nonspecific immunohistochemical marker of core formation in muscle. J. Neurol. Sci. 2003, 206, 71–78. [Google Scholar] [CrossRef]
- van der Flier, A.; Kuikman, I.; Kramer, D.; Geerts, D.; Kreft, M.; Takafuta, T.; Shapiro, S.S.; Sonnenberg, A. Different splice variants of filamin-B affect myogenesis, subcellular distribution, and determine binding to integrin [beta] subunits. J. Cell Biol. 2002, 156, 361–376. [Google Scholar] [CrossRef]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol. Cell Biol. 2006, 26, 6522–6534. [Google Scholar] [CrossRef]
- Juo, L.Y.; Liao, W.C.; Shih, Y.L.; Yang, B.Y.; Liu, A.B.; Yan, Y.T. HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Cui, C.; Zhao, X.; Zhang, Y.; Zhang, Y.; Zhao, J.; Shen, X.; He, H.; Wang, J.; Ma, M.; et al. Filamin C regulates skeletal muscle atrophy by stabilizing dishevelled-2 to inhibit autophagy and mitophagy. Mol. Ther. Nucleic Acids 2022, 27, 147–164. [Google Scholar] [CrossRef]
- Beatham, J.; Romero, R.; Townsend, S.K.; Hacker, T.; van der Ven, P.F.; Blanco, G. Filamin C interacts with the muscular dystrophy KY protein and is abnormally distributed in mouse KY deficient muscle fibres. Hum. Mol. Genet. 2004, 13, 2863–2874. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; Oorschot, V.; Ramm, G.; Bryson-Richardson, R.J. FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Hum. Mol. Genet. 2016, 25, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Jayo, A.; Parsons, M. Fascin: A key regulator of cytoskeletal dynamics. Int. J. Biochem. Cell Biol. 2010, 42, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kim, D.J.; Adams, J.C. The roles of fascins in health and disease. J. Pathol. 2011, 224, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Zanet, J.; Jayo, A.; Plaza, S.; Millard, T.; Parsons, M.; Stramer, B. Fascin promotes filopodia formation independent of its role in actin bundling. J. Cell Biol. 2012, 197, 477–486. [Google Scholar] [CrossRef]
- Ishikawa, R.; Sakamoto, T.; Ando, T.; Higashi-Fujime, S.; Kohama, K. Polarized actin bundles formed by human fascin-1: Their sliding and disassembly on myosin II and myosin V in vitro. J. Neurochem. 2003, 87, 676–685. [Google Scholar] [CrossRef]
- Adams, J.C.; Clelland, J.D.; Collett, G.D.; Matsumura, F.; Yamashiro, S.; Zhang, L. Cell-matrix adhesions differentially regulate fascin phosphorylation. Mol. Biol. Cell 1999, 10, 4177–4190. [Google Scholar] [CrossRef]
- Camuglia, J.M.; Mandigo, T.R.; Moschella, R.; Mark, J.; Hudson, C.H.; Sheen, D.; Folker, E.S. An RNAi based screen in Drosophila larvae identifies fascin as a regulator of myoblast fusion and myotendinous junction structure. Skelet. Muscle 2018, 8, 12. [Google Scholar] [CrossRef]
- Erickson, M.R.; Galletta, B.J.; Abmayr, S.M. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J. Cell Biol. 1997, 138, 589–603. [Google Scholar] [CrossRef]
- Chen, E.H.; Olson, E.N. Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev. Cell 2001, 1, 705–715. [Google Scholar] [CrossRef]
- Junghans, D.; Herzog, S. Cnn3 regulates neural tube morphogenesis and neuronal stem cell properties. FEBS J. 2018, 285, 325–338. [Google Scholar] [CrossRef]
- She, Y.; Li, C.; Jiang, T.; Lei, S.; Zhou, S.; Shi, H.; Chen, R. Knockdown of CNN3 Impairs Myoblast Proliferation, Differentiation, and Protein Synthesis via the mTOR Pathway. Front. Physiol. 2021, 12, 659272. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Liang, R.; Zhao, S.; Wang, R.; Huang, R.; Li, K. CNN3 is regulated by microRNA-1 during muscle development in pigs. Int. J. Biol. Sci. 2014, 10, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, M.; Marampon, F.; Zani, B.M.; Prudente, S.; Perlas, E.; Caputo, V.; Cianetti, L.; Berno, V.; Narumiya, S.; Kang, S.W.; et al. ROCK2 and its alternatively spliced isoform ROCK2m positively control the maturation of the myogenic program. Mol. Cell Biol. 2007, 27, 6163–6176. [Google Scholar] [CrossRef]
- Shibukawa, Y.; Yamazaki, N.; Daimon, E.; Wada, Y. Rock-dependent calponin 3 phosphorylation regulates myoblast fusion. Exp. Cell Res. 2013, 319, 633–648. [Google Scholar] [CrossRef]
- Mancini, A.; Sirabella, D.; Zhang, W.; Yamazaki, H.; Shirao, T.; Krauss, R.S. Regulation of myotube formation by the actin-binding factor drebrin. Skelet. Muscle 2011, 1, 36. [Google Scholar] [CrossRef]
- Stiber, J.A.; Zhang, Z.S.; Burch, J.; Eu, J.P.; Zhang, S.; Truskey, G.A.; Seth, M.; Yamaguchi, N.; Meissner, G.; Shah, R.; et al. Mice lacking Homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol. Cell Biol. 2008, 28, 2637–2647. [Google Scholar] [CrossRef]
- Salanova, M.; Volpe, P.; Blottner, D. Homer protein family regulation in skeletal muscle and neuromuscular adaptation. IUBMB Life 2013, 65, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi-Yamaguchi, Y.; Sato, Y.; Sakai, R.; Mizutani, A.; Knopfel, T.; Mori, N.; Mikoshiba, K.; Furuichi, T. Interaction of Cupidin/Homer2 with two actin cytoskeletal regulators, Cdc42 small GTPase and Drebrin, in dendritic spines. BMC Neurosci. 2009, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, S.; Khanzada, U.K.; Parsons, M.; Chilton, J.K.; Gordon-Weeks, P.R. Targeting of the F-actin-binding protein drebrin by the microtubule plus-tip protein EB3 is required for neuritogenesis. Nat. Cell Biol. 2008, 10, 1181–1189. [Google Scholar] [CrossRef]
- Straube, A.; Merdes, A. EB3 regulates microtubule dynamics at the cell cortex and is required for myoblast elongation and fusion. Curr. Biol. 2007, 17, 1318–1325. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Nissar, A.A.; Al-Sajee, D.; Tarnopolsky, M.A.; Parise, G.; Lach, B.; Furst, D.O.; van der Ven, P.F.M.; Kley, R.A.; Hawke, T.J. Xin is a marker of skeletal muscle damage severity in myopathies. Am. J. Pathol. 2013, 183, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, S.C.; Hawke, T.J.; Gallardo, T.D.; Richardson, J.A.; Garry, D.J. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol. Genom. 2003, 14, 261–271. [Google Scholar] [CrossRef]
- Nissar, A.A.; Zemanek, B.; Labatia, R.; Atkinson, D.J.; van der Ven, P.F.; Furst, D.O.; Hawke, T.J. Skeletal muscle regeneration is delayed by reduction in Xin expression: Consequence of impaired satellite cell activation? Am. J. Physiol. Cell Physiol. 2012, 302, C220–C227. [Google Scholar] [CrossRef] [PubMed]
- Hawke, T.J.; Atkinson, D.J.; Kanatous, S.B.; Van der Ven, P.F.; Goetsch, S.C.; Garry, D.J. Xin, an actin binding protein, is expressed within muscle satellite cells and newly regenerated skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 2007, 293, C1636–C1644. [Google Scholar] [CrossRef]
- van der Ven, P.F.; Ehler, E.; Vakeel, P.; Eulitz, S.; Schenk, J.A.; Milting, H.; Micheel, B.; Furst, D.O. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp. Cell Res. 2006, 312, 2154–2167. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, S.C.; Martin, C.M.; Embree, L.J.; Garry, D.J. Myogenic progenitor cells express filamin C in developing and regenerating skeletal muscle. Stem Cells Dev. 2005, 14, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Hassel, D.; Dahme, T.; Erdmann, J.; Meder, B.; Huge, A.; Stoll, M.; Just, S.; Hess, A.; Ehlermann, P.; Weichenhan, D.; et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 2009, 15, 1281–1288. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Han, Y.; Chen, J.; Wang, Y.; Zhang, Z.; Hui, R. NELIN, a new F-actin associated protein, stimulates HeLa cell migration and adhesion. Biochem. Biophys. Res. Commun. 2005, 330, 1127–1131. [Google Scholar] [CrossRef]
- Mahmassani, Z.S.; Reidy, P.T.; McKenzie, A.I.; Stubben, C.; Howard, M.T.; Drummond, M.J. Age-dependent skeletal muscle transcriptome response to bed rest-induced atrophy. J. Appl. Physiol. 2019, 126, 894–902. [Google Scholar] [CrossRef]
- Kostek, M.C.; Chen, Y.W.; Cuthbertson, D.J.; Shi, R.; Fedele, M.J.; Esser, K.A.; Rennie, M.J. Gene expression responses over 24 h to lengthening and shortening contractions in human muscle: Major changes in CSRP3, MUSTN1, SIX1, and FBXO32. Physiol. Genom. 2007, 31, 42–52. [Google Scholar] [CrossRef]
- Lee, A.; Hakuno, F.; Northcott, P.; Pessin, J.E.; Rozakis Adcock, M. Nexilin, a cardiomyopathy-associated F-actin binding protein, binds and regulates IRS1 signaling in skeletal muscle cells. PLoS ONE 2013, 8, e55634. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.; Gross, S.M.; David, L.L.; Klimek, J.E.; Rotwein, P. Separating myoblast differentiation from muscle cell fusion using IGF-I and the p38 MAP kinase inhibitor SB202190. Am. J. Physiol. Cell Physiol. 2015, 309, C491–C500. [Google Scholar] [CrossRef]
- Hung, R.J.; Spaeth, C.S.; Yesilyurt, H.G.; Terman, J.R. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 2013, 15, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Beuchle, D.; Schwarz, H.; Langegger, M.; Koch, I.; Aberle, H. Drosophila MICAL regulates myofilament organization and synaptic structure. Mech. Dev. 2007, 124, 390–406. [Google Scholar] [CrossRef]
- Giarratana, N.; Conti, F.; La Rovere, R.; Gijsbers, R.; Carai, P.; Duelen, R.; Vervliet, T.; Bultynck, G.; Ronzoni, F.; Piciotti, R.; et al. MICAL2 is essential for myogenic lineage commitment. Cell Death Dis. 2020, 11, 654. [Google Scholar] [CrossRef]
- Marotta, M.; Ruiz-Roig, C.; Sarria, Y.; Peiro, J.L.; Nunez, F.; Ceron, J.; Munell, F.; Roig-Quilis, M. Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol. Genom. 2009, 37, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.; Mackels, L.; Markati, T.; Sarkozy, A.; Ochala, J.; Jungbluth, H.; Ramdas, S.; Servais, L. Early clinical and pre-clinical therapy development in Nemaline myopathy. Expert. Opin. Ther. Targets 2022, 26, 853–867. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhou, A.; Nguyen, D.T.; Zhang, H.S.; Benz, E.J., Jr. Protein 4.1R Influences Myogenin Protein Stability and Skeletal Muscle Differentiation. J. Biol. Chem. 2016, 291, 25591–25607. [Google Scholar] [CrossRef]
- Swailes, N.T.; Colegrave, M.; Knight, P.J.; Peckham, M. Non-muscle myosins 2A and 2B drive changes in cell morphology that occur as myoblasts align and fuse. J. Cell Sci. 2006, 119, 3561–3570. [Google Scholar] [CrossRef]
- Myhre, J.L.; Pilgrim, D.B. At the Start of the Sarcomere: A Previously Unrecognized Role for Myosin Chaperones and Associated Proteins during Early Myofibrillogenesis. Biochem. Res. Int. 2012, 2012, 712315. [Google Scholar] [CrossRef]
- Bang, M.L.; Li, X.; Littlefield, R.; Bremner, S.; Thor, A.; Knowlton, K.U.; Lieber, R.L.; Chen, J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J. Cell Biol. 2006, 173, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Vlahovich, N.; Schevzov, G.; Nair-Shaliker, V.; Ilkovski, B.; Artap, S.T.; Joya, J.E.; Kee, A.J.; North, K.N.; Gunning, P.W.; Hardeman, E.C. Tropomyosin 4 defines novel filaments in skeletal muscle associated with muscle remodelling/regeneration in normal and diseased muscle. Cell Motil. Cytoskelet. 2008, 65, 73–85. [Google Scholar] [CrossRef]
- Michele, D.E.; Metzger, J.M. Physiological consequences of tropomyosin mutations associated with cardiac and skeletal myopathies. J. Mol. Med. 2000, 78, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, I.; Alexander, M.S.; Kunkel, L.M. miRNAS in normal and diseased skeletal muscle. J. Cell Mol. Med. 2009, 13, 2–11. [Google Scholar] [CrossRef]
- Ouyang, Z.; Wei, K. miRNA in cardiac development and regeneration. Cell Regen. 2021, 10, 14. [Google Scholar] [CrossRef]
- Jin, Z.Q. MicroRNA targets and biomarker validation for diabetes-associated cardiac fibrosis. Pharmacol. Res. 2021, 174, 105941. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Catellani, C.; Sartori, C.; Lazzeroni, P.; Amarri, S.; Street, M.E. Obesity, Insulin Resistance, and Colorectal Cancer: Could miRNA Dysregulation Play A Role? Int. J. Mol. Sci. 2019, 20, 2922. [Google Scholar] [CrossRef]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Guo, Y.; Liu, X.; Song, Y.; Li, X.; Ding, X.; Guo, H. A novel lncRNA promotes myogenesis of bovine skeletal muscle satellite cells via PFN1-RhoA/Rac1. J. Cell Mol. Med. 2021, 25, 5988–6005. [Google Scholar] [CrossRef]
- Mishima, Y.; Abreu-Goodger, C.; Staton, A.A.; Stahlhut, C.; Shou, C.; Cheng, C.; Gerstein, M.; Enright, A.J.; Giraldez, A.J. Zebrafish miR-1 and miR-133 shape muscle gene expression and regulate sarcomeric actin organization. Genes. Dev. 2009, 23, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, Y.; Wang, Z.; Zhao, S.; Mu, Y.; Li, K. Integrated analysis of miRNA and mRNA paired expression profiling of prenatal skeletal muscle development in three genotype pigs. Sci. Rep. 2015, 5, 15544. [Google Scholar] [CrossRef] [PubMed]
- Mok, G.F.; Lozano-Velasco, E.; Münsterberg, A. microRNAs in skeletal muscle development. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2017; pp. 67–76. [Google Scholar]
- Holstein, I.; Singh, A.K.; Pohl, F.; Misiak, D.; Braun, J.; Leitner, L.; Huttelmaier, S.; Posern, G. Post-transcriptional regulation of MRTF-A by miRNAs during myogenic differentiation of myoblasts. Nucleic Acids Res. 2020, 48, 8927–8942. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Kuwahara, K.; Nakagawa, Y.; Takaoka, M.; Kinoshita, H.; Nakao, K.; Kuwabara, Y.; Yamada, Y.; Yamada, C.; Shibata, J.; et al. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice. EMBO J. 2012, 31, 4428–4440. [Google Scholar] [CrossRef]
- Zhuang, C.; Yuan, Y.; Song, T.; Wang, H.; Huang, L.; Luo, X.; He, H.; Huo, L.; Zhou, H.; Wang, N.; et al. miR-219a-5p inhibits breast cancer cell migration and epithelial-mesenchymal transition by targeting myocardin-related transcription factor A. Acta Biochim. Biophys. Sin. 2017, 49, 1112–1121. [Google Scholar] [CrossRef]
- Tan, D.X.; Chen, X.X.; Bai, T.Z.; Zhang, J.; Li, Z.F. Sevoflurane up-regulates microRNA-204 to ameliorate myocardial ischemia/reperfusion injury in mice by suppressing Cotl1. Life Sci. 2020, 259, 118162. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Lee, W. MiR-320-3p Regulates the Proliferation and Differentiation of Myogenic Progenitor Cells by Modulating Actin Remodeling. Int. J. Mol. Sci. 2022, 23, 801. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Min, K.H.; Lee, W. Palmitic Acid-Induced miR-429-3p Impairs Myoblast Differentiation by Downregulating CFL2. Int. J. Mol. Sci. 2021, 22, 10972. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Lee, W. Role of MiR-325-3p in the Regulation of CFL2 and Myogenic Differentiation of C2C12 Myoblasts. Cells 2021, 10, 2725. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Lee, W. MiR-141-3p regulates myogenic differentiation in C2C12 myoblasts via CFL2-YAP-mediated mechanotransduction. BMB Rep. 2022, 55, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Fariyike, B.; Singleton, Q.; Hunter, M.; Hill, W.D.; Isales, C.M.; Hamrick, M.W.; Fulzele, S. Role of MicroRNA-141 in the Aging Musculoskeletal System: A Current Overview. Mech. Ageing Dev. 2019, 178, 9–15. [Google Scholar] [CrossRef]
- Chang, S.Y.; Han, S.Z.; Choe, H.M.; Gao, K.; Jin, Z.Y.; Liu, X.Y.; Yang, L.H.; Lv, S.T.; Yin, X.J.; Quan, L.H.; et al. miR-320 regulates myogenesis by targeting growth factor receptor-bound protein-2 and ameliorates myotubes atrophy. Int. J. Biochem. Cell Biol. 2022, 147, 106212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.R.; Zhang, M.; Li, F.; Li, D.H.; Sun, G.R.; Liu, X.J.; Li, H.; Han, R.L.; Jiang, R.R.; Li, Z.J.; et al. Study on the role of gga-miRNA-200a in regulating cell differentiation and proliferation of chicken breast muscle by targeting Grb2. Anim. Cells Syst. 2017, 21, 365–373. [Google Scholar] [CrossRef]
- Shen, X.; Tang, J.; Jiang, R.; Wang, X.; Yang, Z.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. CircRILPL1 promotes muscle proliferation and differentiation via binding miR-145 to activate IGF1R/PI3K/AKT pathway. Cell Death Dis. 2021, 12, 142. [Google Scholar] [CrossRef]
- Du, J.; Zhang, Y.; Shen, L.; Luo, J.; Lei, H.; Zhang, P.; Pu, Q.; Liu, Y.; Shuai, S.; Li, Q.; et al. Effect of miR-143-3p on C2C12 myoblast differentiation. Biosci. Biotechnol. Biochem. 2016, 80, 706–711. [Google Scholar] [CrossRef]
- Zhang, W.R.; Zhang, H.N.; Wang, Y.M.; Dai, Y.; Liu, X.F.; Li, X.; Ding, X.B.; Guo, H. miR-143 regulates proliferation and differentiation of bovine skeletal muscle satellite cells by targeting IGFBP5. Vitr. Cell Dev. Biol. Anim. 2017, 53, 265–271. [Google Scholar] [CrossRef]
- Zuo, J.; Wu, F.; Liu, Y.; Xiao, J.; Xu, M.; Yu, Q.; Xia, M.; He, X.; Zou, S.; Tan, H.; et al. MicroRNA Transcriptome Profile Analysis in Porcine Muscle and the Effect of miR-143 on the MYH7 Gene and Protein. PLoS ONE 2015, 10, e0124873. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, B.; Zhao, Y.; Raza, S.H.A.; Li, Y.; Wang, J.; Ma, X.; Almohaimeed, H.M.; Shaheen, S.; Al-Sarraj, F.; et al. Proliferation of bovine myoblast by LncPRRX1 via regulation of the miR-137/CDC42 axis. Int. J. Biol. Macromol. 2022, 220, 33–42. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes. Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef]


| Protein Groups | ABPs | Expression | Function | Mechanism | Refs. |
|---|---|---|---|---|---|
| Proteins regulating F-actin assembly | PFN1 | Expressed early in muscle development | Negatively regulates myogenic differentiation. | Represses RhoA and Rac1 at transcriptional levels | [76,77] |
| Binds to Cdc42 activity, activates PAK/JNK signaling pathways | [81] | ||||
| PFN2 | Expressed early in development | Inhibits cell proliferation while inducing apoptosis, represses the myogenic differentiation of C2C12 myoblasts | Reduces HDAC1 localization and subsequently induces p53 Expression | [83] | |
| Proteins regulating F-actin disassembly | CFL1 | Expressed in the early stage of differentiation and declined during muscle development | Overexpression of CFL1 suppresses the differentiation of bovine primary myoblasts. | Increases actin depolymerization | [89] |
| CFL2 | Expressed later and increases during muscle differentiation and fusion | CFL2-knockout mice lead to an abnormal accumulation of F-actin and progressive disturbance of the sarcomeric architecture of skeletal muscles. | Decreases p38 MAPK, CBP, AMPKα1, and MEF2C pathway and increases ERK2 expression | [93,94,95,96] | |
| CFL2 knockdown in C2C12 myoblast cells inhibits myogenic differentiation and promotes cell proliferation. | Abnormal F-actin formation modulates nuclear YAP localization | [26,27] | |||
| WDR1 | Highly expressed during exercise after muscle disuse | WDR1-knockout mice cause postnatal lethality, sarcomere disorganization, and contractility defects. | Actin aggregate formation | [81,97] | |
| CAP1 | Downregulated during myogenic differentiation. | CAP1 deletion results in a spread-out morphology, increased cell size and nuclei, and inversely correlating with myogenic differentiation. | Regulates F-actin organization Regulate the expression of myoblast profusion molecules, such as β1D-integrin, Caveolin-3, Myomaker, and Myomixe. | [100] | |
| CAP2 | Increased upon differentiation in vitro or regeneration in vivo | Knockout CAP1 in mice is characterized by delayed maturation of motor functions, reduced muscle strength, and weakness. | α-SMA and α-CAA, an internal piece of sarcomere F-actin accumulates, thereby leading to ring fibers | [28] | |
| TWF1 | Increased in the early stage of myoblast fusion, then gradually decreased | Its knockdown promotes cell proliferation and inhibits myogenic differentiation. | TWF1 knockdown accumulates F-actin, leading to nuclear YAP localization | [81] | |
| Monomer-sequestering proteins | Tb4 | Upregulated during myotube differentiation of C2C12 cells | Promotes new myofiber formation | Intracellular G-actin-sequestering and inhibits the assembly of actin fibers | [111,112] |
| MRTF-A | Expressed in the early stage of differentiation and declined during differentiation | MRTF-A promotes differentiation of myoblasts and the expression of MyoD and MyoG | Rho/MRTF/SRF pathway and regulate Pax7 expression | [117,118,127] | |
| Proteins for nucleation sites in actin branching | Arp2/3 | Expressed in skeletal muscles | Mutants in Arp2/3 have a fusion block and foci phenotype | Inhibits actin polymerization in myoblast fusion | [129] |
| N-WASP | Expressed in the early phase of mouse embryonic development | Knockout of N-WASP causes early embryonic lethality, characterized by developmental delay | Inhibits the formation of filopodia and lamellipodia, which are required for cell movement. | [141] | |
| Knockout N-WASP in myoblasts fail to fuse and form multinucleated myotubes | [136,137] | ||||
| WAVE2 | Concentrated in the leading edges of lamellipodia in myoblasts | Promotes myoblast fusion, promotes lamellipodial formation and subsequent migration in C2C12 cells | Acts downstream of HGF/PI3K | [29,30] | |
| Fhod3 | Accumulated in skeletal muscles | Flight muscle myofibrils were disrupted in muscle-specific silencing of Fhos in Drosophila. | Sarcomere organization | [151,152] | |
| DAAM1 | Accumulated in skeletal muscles | Absence of DAAM1 results in disorganization of thin filaments, leading to shorter and sparsely distributed sarcomeres. | Regulated F-actin content | [29,31] | |
| Dia | Accumulated in skeletal muscles | Regulating the length and width of each sarcomere during flight muscle development, required during myoblast fusion | Regulated F-actin content | [136,154] | |
| CTTN | Expressed in skeletal muscles | Facilitates GLUT4myc translocation in L6 myotubes and C2C12 cells | F-actin and stress fiber acculation | [168,169] | |
| Actin nucleation activators | Cdc42/Rac | Expressed in skeletal muscles | Cdc42/Rac mutated embryonic mice exhibited thinner muscle fibers and myoblast fusion. | Regulates the expression of F-actin/Arp2/3 complex | [142] |
| Grb2 | Expressed in skeletal muscles | Grb2 mediates the inhibition of myogenic differentiation by enhancing actin polymerization. | Regulates N-WASP by direct binding | [137] | |
| Toca-1 | Expressed in skeletal muscles | Reduction of Toca-1 in C2C12 cells leads to a substantial decrease in myogenic differentiation. | Regulates N-WASP | [148] | |
| Capping proteins | Gel | Induced steadily during myoblast differentiation | Limits myoblast fusion in Drosophila | Binding to F-actin in the barbed end | [175] |
| CapZ | Upregulated in myotubes | Overexpressed CapZ-β2 results in severe myofibril disruption and, ultimately, death | Sarcomere organization | [177] | |
| Tmod1 | Decreased during differentiation | Significantly inhibits myogenic differentiation | Reducing the expression of muscle-specific genes | [136,183,185] | |
| Tmod4 | Decreased during differentiation | Significantly inhibits myogenic differentiation | MEF2A can bind to the promoter region of Tmod4 and inhibit Tmod4 expression | [182] | |
| Lmod3 | Induced and promotes myoblast differentiation | Loss of Lmod3 function leads to lethal nemaline myopathy. | Sarcomere organization and regulated by MRTF/SRF and MEF2 | [190] | |
| Actin filament cross-linking proteins | Flnc | Expressed early and decreases during cell differentiation | Regulates skeletal muscle development. Flnc-knockdown cells decreases myoblast fusion and rounded fibers. | Associated with the muscle-specific protein HSPB7 or disheveled-2 (Dvl2), which induces the wnt/β-catenin signaling pathway | [29,197,198,199] |
| Fascin | Increased during differentiation | Depletion of fascin disrupts MTJ integrity | Slide as fast as single actin filaments on myosin II and myosin V | [129,205,207,208,209] | |
| F-actin stabilizing proteins | CNN3 | Increased during muscle development and regeneration in pigs and mice | Knockdown CNN3 dramatically suppresses differentiation. | Akt/mTOR and AMPK/mTOR, ROCK | [211,214] |
| Dbn1 | Increases during myogenic differentiation | Inhibition of Dbn1-induced cell proliferation or apoptosis, suppresses myoblast differentiation. | p38 MAPK-dependent manner, Homer/Cdc42, EB3 | [215,216,217,218,220] | |
| Xin | Upregulated during the early phases of skeletal muscle regeneration | Reduced proliferation and apoptosis, and promotes myogenic differentiation | Works with other partners, such as Ena/VASP homology 1 and Flnc | [224] | |
| NEXN | Highly expressed in the heart and skeletal muscles. Increased in fast-growth compared to slow-growth chicken muscle | Promotes glucose uptake, and myotube formation in L6 cells | IRS1/NEXN complex negatively regulates the IRS1/PI3K/Akt signaling pathway | [229] | |
| Promotes myotube fusion C2 myoblasts | Association IGF-I/p38α/β/kinase-mediated signaling | [232] | |||
| MICAL2 | A significant increase in myogenic differentiation and skeletal muscle regeneration | Promotes degeneration and regeneration | Redox alteration of nuclear actin and associated activation of myogenic transcription factors | [236] | |
| Muscle contractile-related ABPs in myogenic differentiation | α-Actinin | Gradually increased during the differentiation | Links actin filaments at the Z-discs, imparting structural reinforcement and serving as an anchor for the thin filaments | Organizing and stabilizing the sarcomere | [238] |
| NM II-A and -II-B | Expressed in fully differentiated myofiber | Assists the fusion of newly generated myoblasts | [239,240] | ||
| Nebulin | Expressed in skeletal muscle | Interact with individual monomers of the actin filaments | [70,241] | ||
| Tpms | Accumulated in skeletal muscles | Regulate muscle remodeling/regeneration | [242,243] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Dash, R.; Jeong, K.; Lee, W. Role of Actin-Binding Proteins in Skeletal Myogenesis. Cells 2023, 12, 2523. https://doi.org/10.3390/cells12212523
Nguyen MT, Dash R, Jeong K, Lee W. Role of Actin-Binding Proteins in Skeletal Myogenesis. Cells. 2023; 12(21):2523. https://doi.org/10.3390/cells12212523
Chicago/Turabian StyleNguyen, Mai Thi, Raju Dash, Kyuho Jeong, and Wan Lee. 2023. "Role of Actin-Binding Proteins in Skeletal Myogenesis" Cells 12, no. 21: 2523. https://doi.org/10.3390/cells12212523
APA StyleNguyen, M. T., Dash, R., Jeong, K., & Lee, W. (2023). Role of Actin-Binding Proteins in Skeletal Myogenesis. Cells, 12(21), 2523. https://doi.org/10.3390/cells12212523