
Proteomic Analyses of the G Protein-Coupled Estrogen Receptor GPER1 Reveal Constitutive Links to Endoplasmic Reticulum, Glycosylation, Trafficking, and Calcium Signaling

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Cloning
2.3. Immunofluorescence Staining and Microscopy
2.4. Rac1 Sensor Assays
2.5. Proximity Labeling Experiments
2.6. Pull-Down of Biotinylated Proteins for MS
2.7. Immunoblot Analysis of Biotinylated Proteins
2.8. IP of F-GPER1 and F-GPER1-APEX2 for MS
2.9. Immunoblot Analysis of Immunoprecipitated Samples
2.10. General Data Analyses
2.11. Proteomics
2.11.1. Preparation of Peptides of Proximity Labeling Samples
2.11.2. LC-MS/MS Analyses of Proximity Labeling Samples
2.11.3. Preparation of Peptides of IP Samples
2.11.4. LC-MS/MS Analyses of IP Samples
3. Results
3.1. Localization of F-GPER1 and Fusion Protein F-GPER1-APEX2
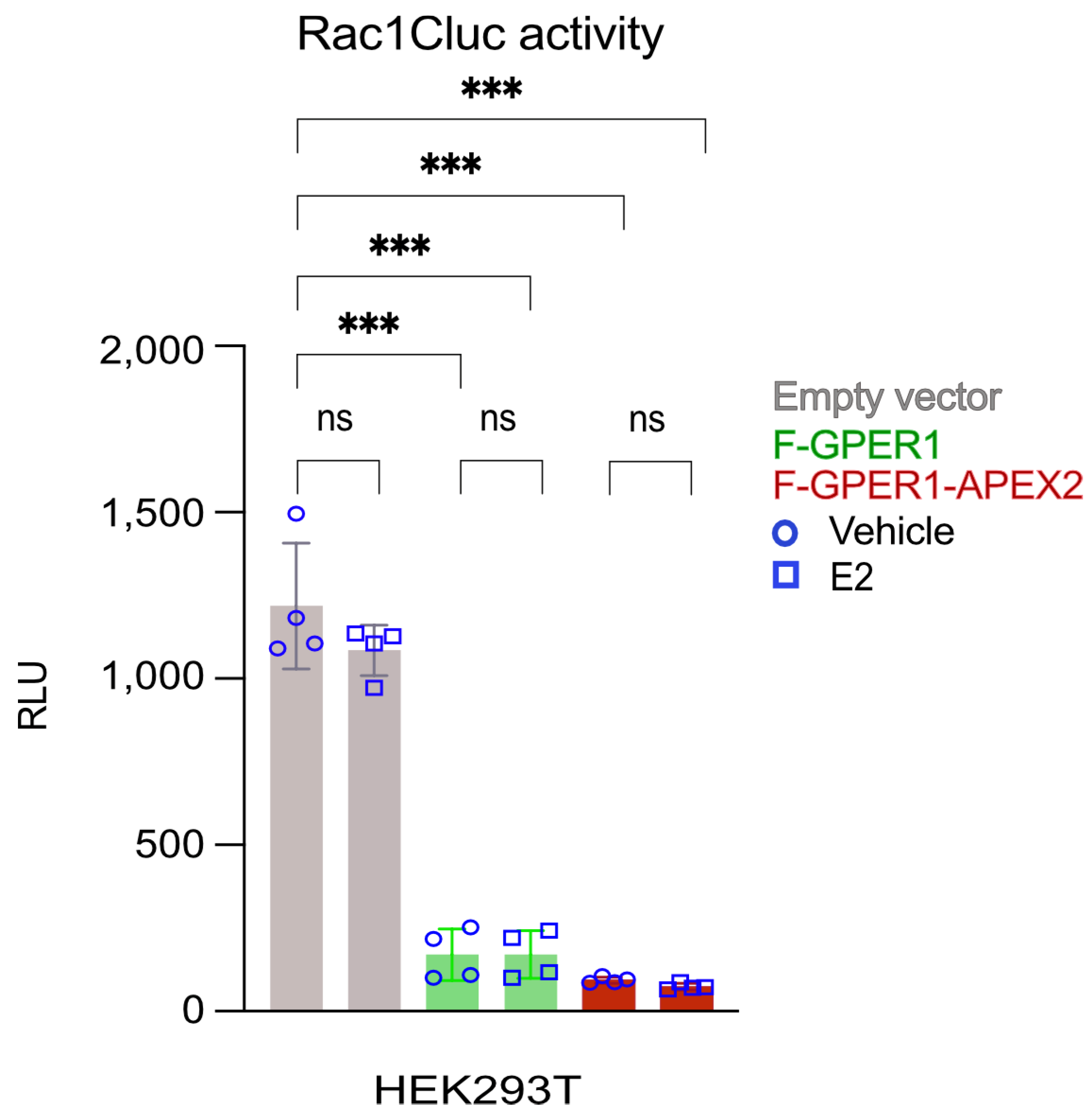
3.2. Conformational Rac1 Sensor Assay Indicates Constitutive F-GPER1 and F-GPER1-APEX2 Activities
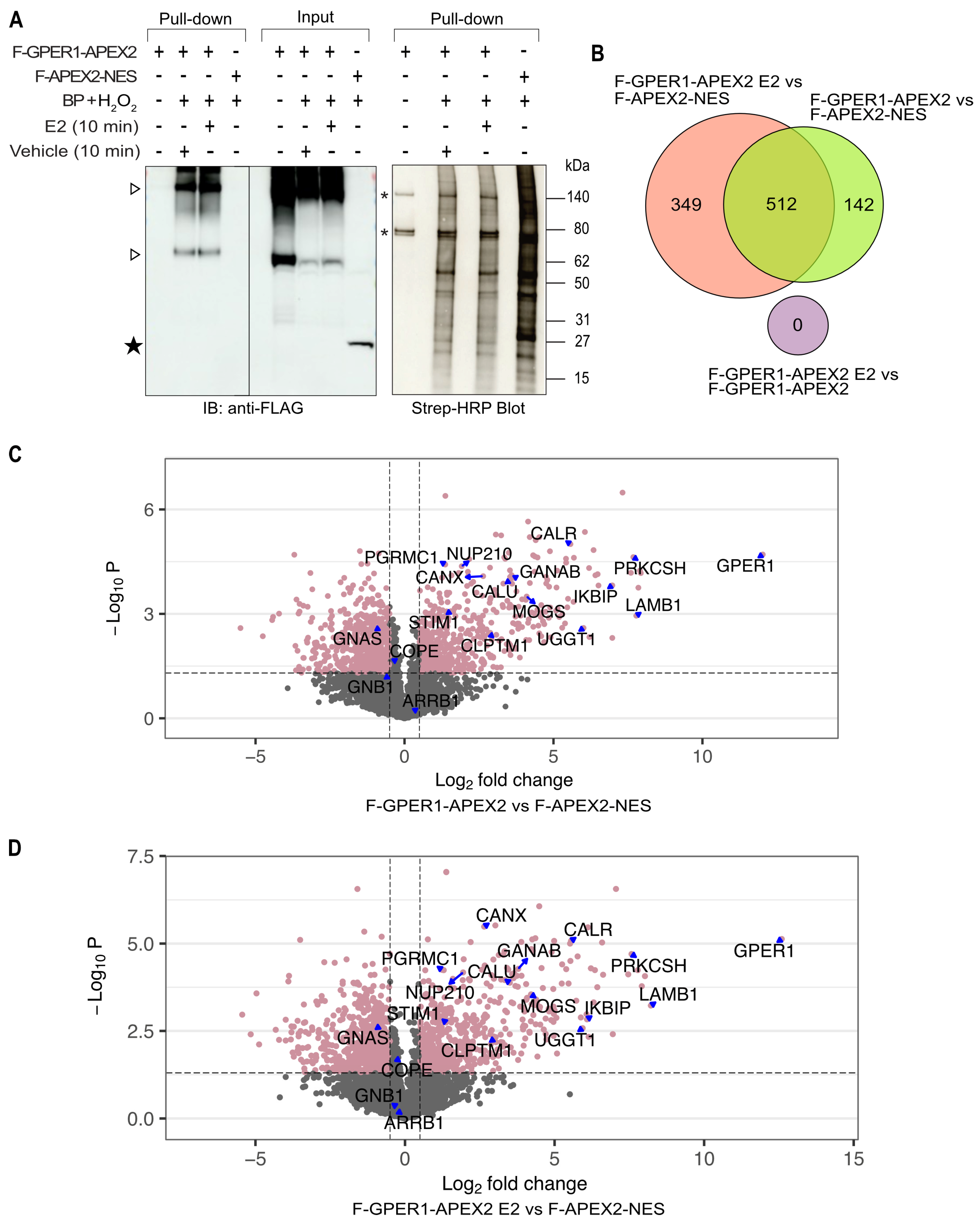
3.3. Identification of Potential GPER1 Interaction Partners by Proximity Labeling
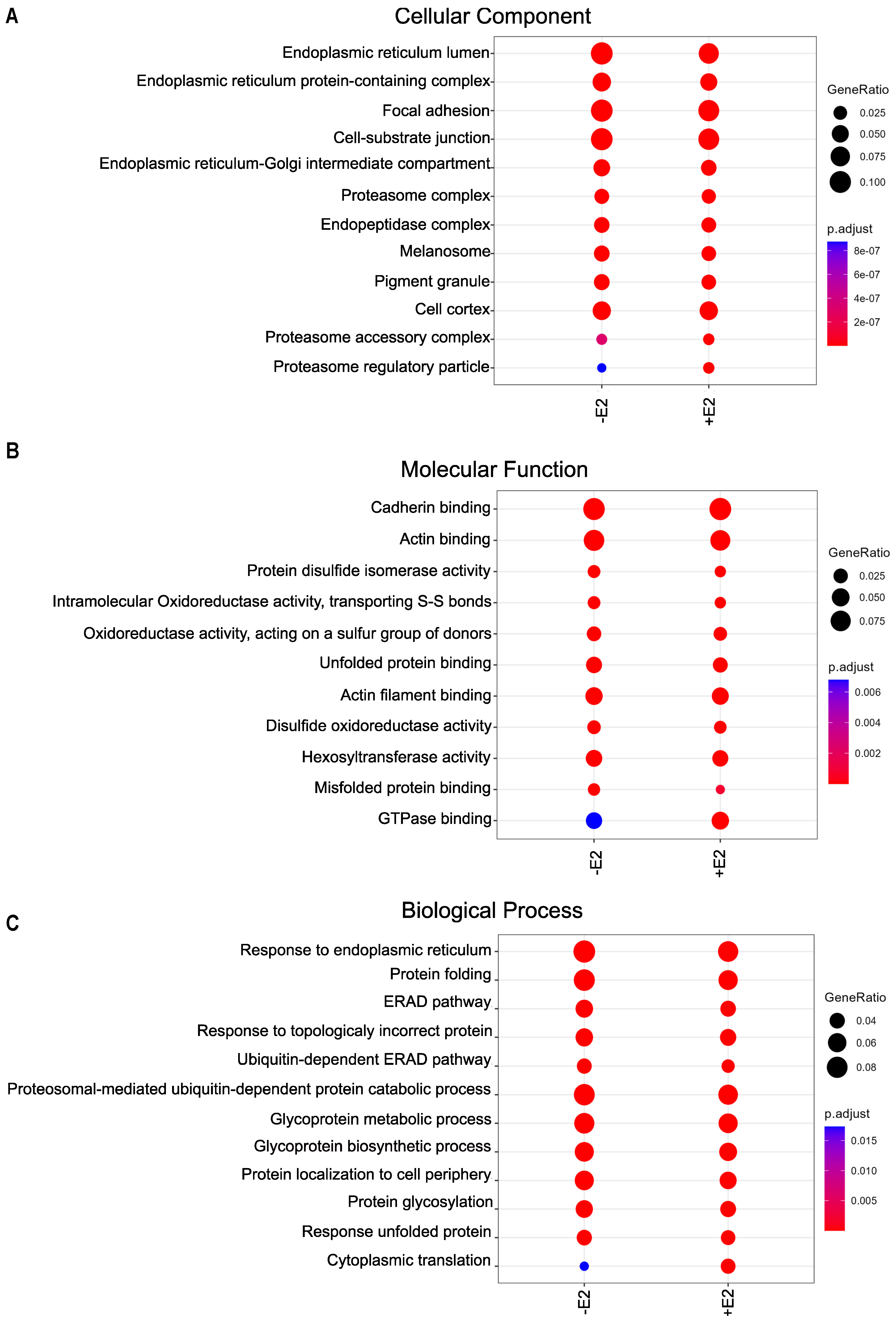
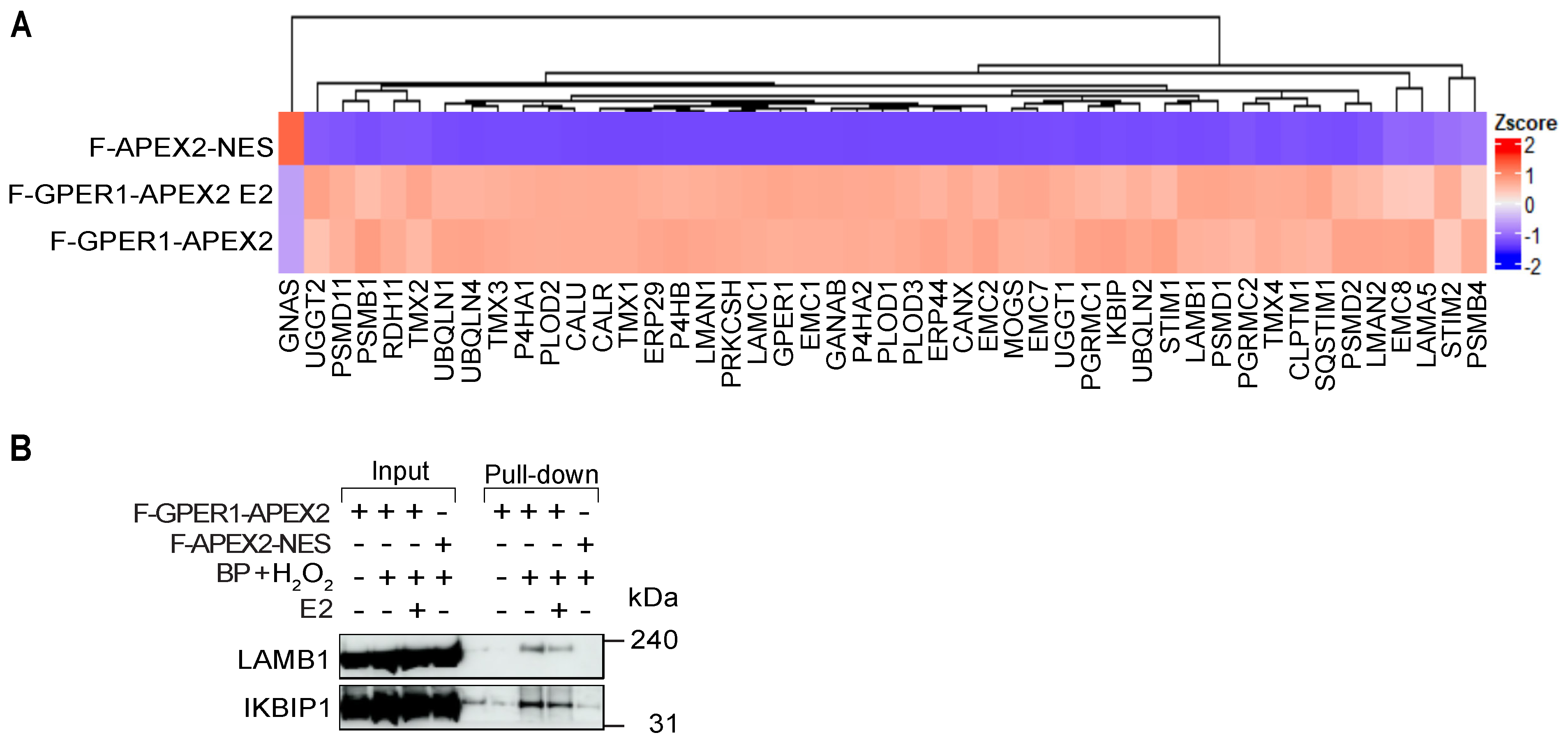
3.4. Wide Variety of Potential Interaction Partners for GPER1
3.5. Identification of Potential GPER1 Interactors by IP-LC-MS/MS
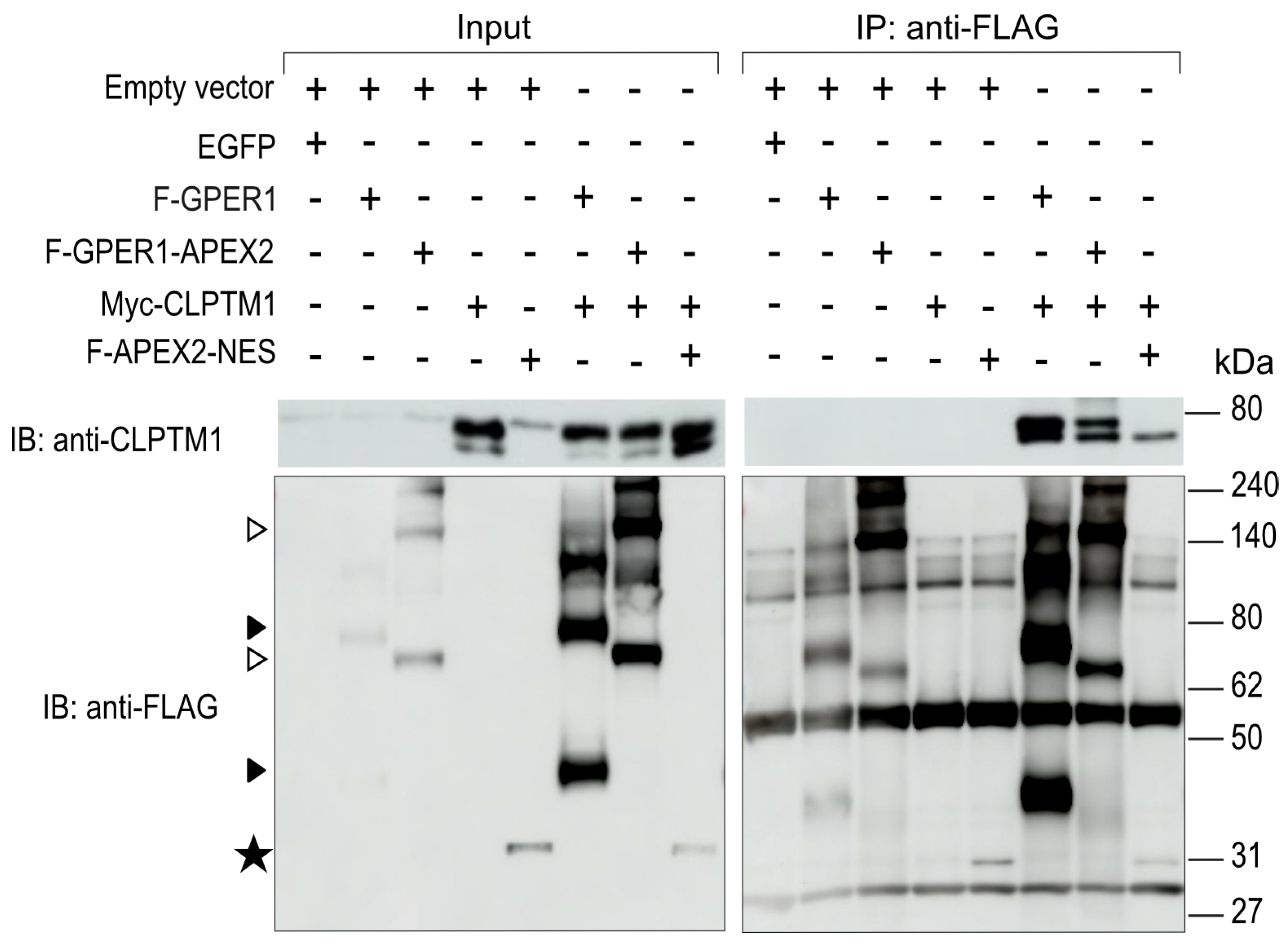
3.6. F-GPER1 Interacts with Myc-CLPTM1 in HEK293T Cells
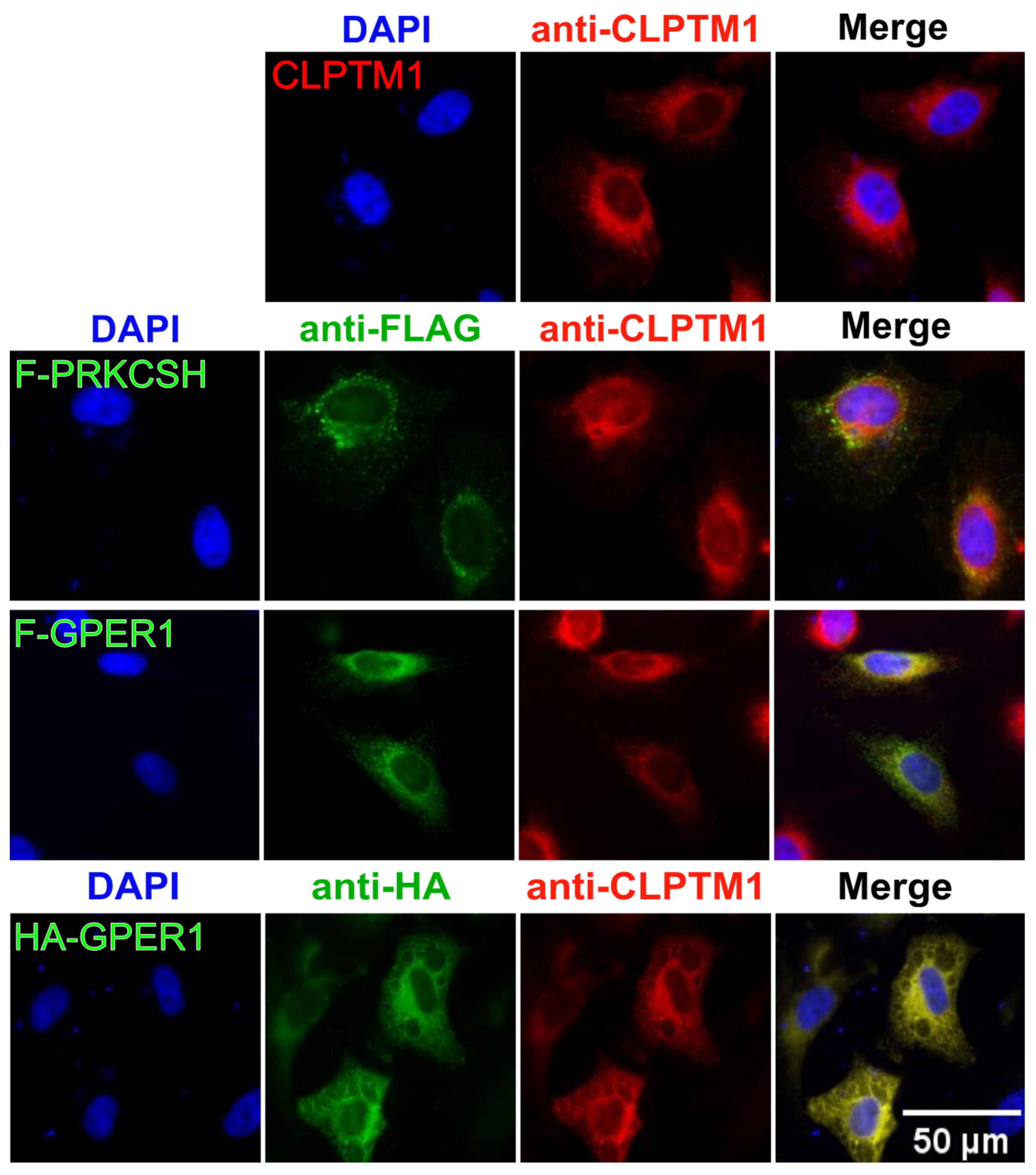
3.7. F-GPER1, CLPTM1, and F-PRKCSH Colocalization and Translocation
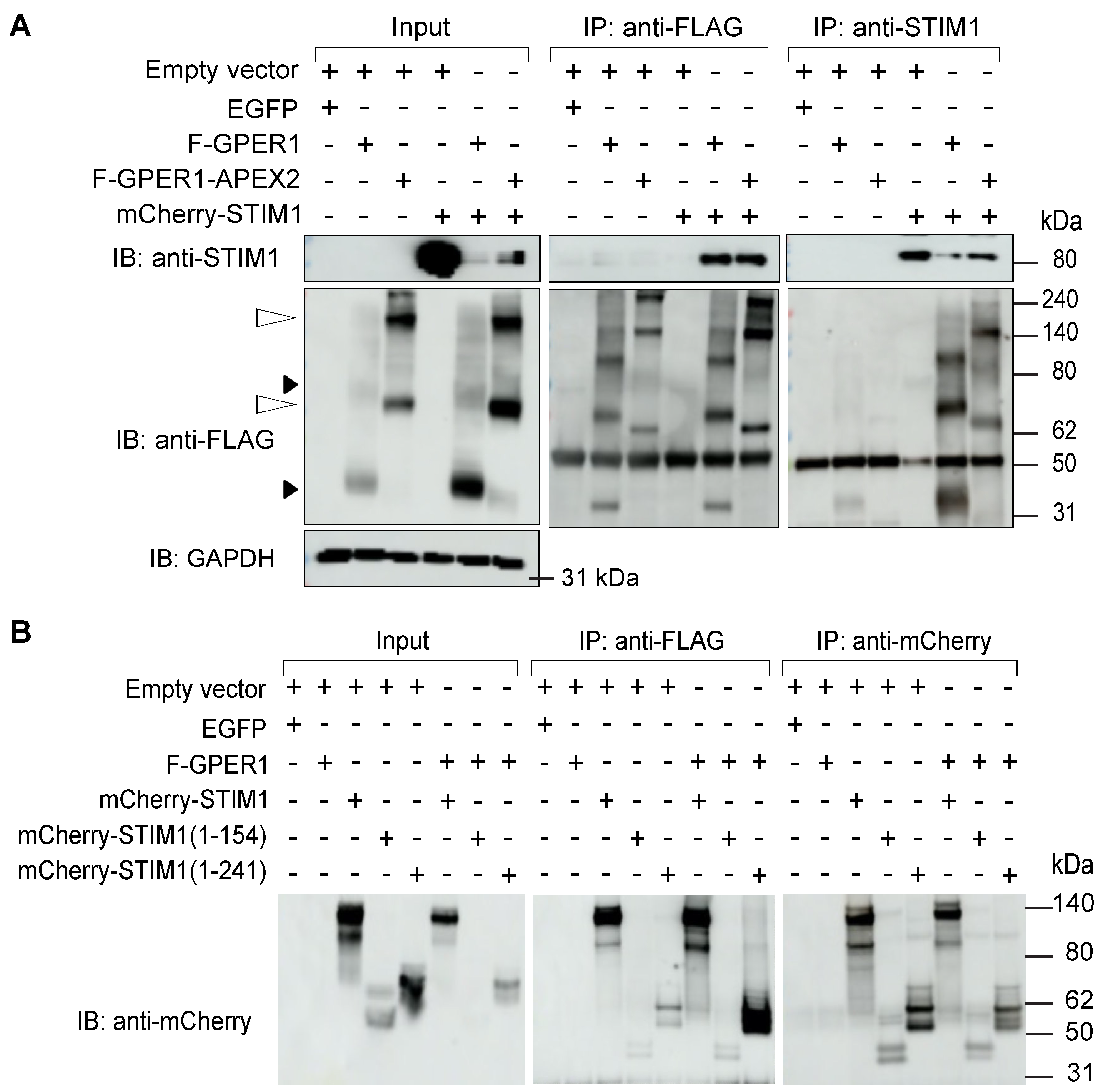
3.8. F-GPER1 Interacts with mCherry-STIM1 in HEK293T Cells
3.9. mCherry-STIM1 and F-GPER1 Affect the Expression Levels of Each Other
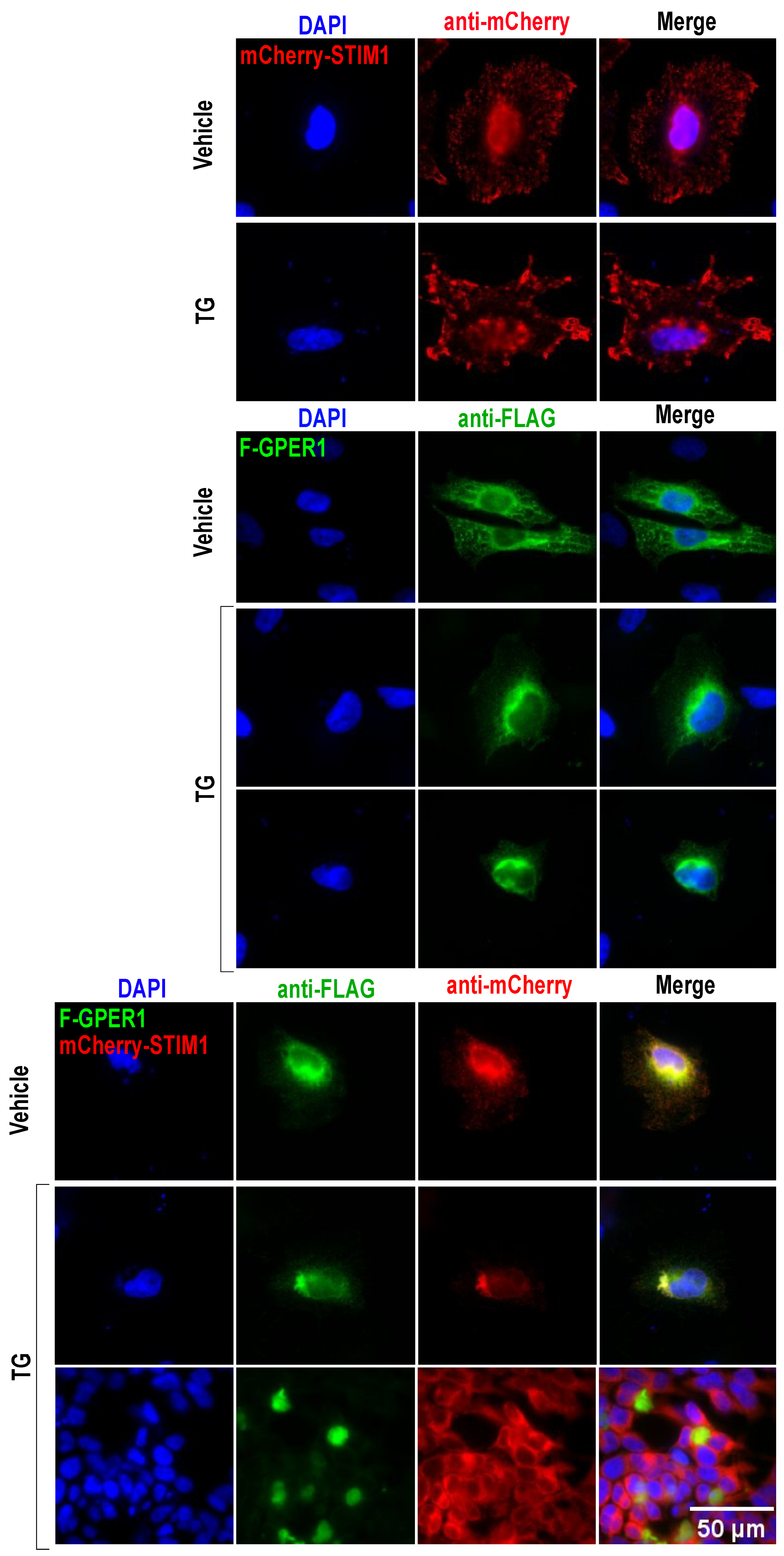
3.10. F-GPER1 and mCherry-STIM1 Colocalization and Translocation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.; Quinn, J.; Pang, Y.; Graeber, C.; Shaw, S.; Dong, J.; Thomas, P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, 3236–3245. [Google Scholar] [CrossRef]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G protein-coupled oestrogen receptor GPER in health and disease: An update. Nat. Rev. Endocrinol. 2023, 19, 407–424. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef]
- Cheng, S.B.; Graeber, C.T.; Quinn, J.A.; Filardo, E.J. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids 2011, 76, 892–896. [Google Scholar] [CrossRef]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Jung, J. Role of G protein-coupled estrogen receptor in cancer progression. Toxicol. Res. 2019, 35, 209–214. [Google Scholar] [CrossRef]
- DeLeon, C.; Wang, D.Q.; Arnatt, C.K. G protein-coupled estrogen receptor, GPER1, offers a novel target for the treatment of digestive diseases. Front. Endocrinol. 2020, 11, 578536. [Google Scholar] [CrossRef] [PubMed]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic perspectives on the modulation of G-protein coupled estrogen receptor, GPER, function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef]
- Yang, S.; Yin, Z.; Zhu, G. A review of the functions of G protein-coupled estrogen receptor 1 in vascular and neurological aging. Eur. J. Pharmacol. 2021, 908, 174363. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef]
- Otto, C.; Rohde-Schulz, B.; Schwarz, G.; Fuchs, I.; Klewer, M.; Brittain, D.; Langer, G.; Bader, B.; Prelle, K.; Nubbemeyer, R.; et al. G protein-coupled receptor 30 localizes to the endoplasmic reticulum and is not activated by estradiol. Endocrinology 2008, 149, 4846–4856. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhang, X.; Xie, Y.; Tu, Y.; Wang, D.; Liu, Z.; Wang, Z.Y. Involvement of estrogen receptor variant ER-α36, not GPR30, in nongenomic estrogen signaling. Mol. Endocrinol. 2010, 24, 709–721. [Google Scholar] [CrossRef]
- Sousa, C.; Ribeiro, M.; Rufino, A.T.; Leitao, A.J.; Mendes, A.F. Assessment of cell line competence for studies of pharmacological GPR30 modulation. J. Recept. Signal Transduct. Res. 2017, 37, 181–188. [Google Scholar] [CrossRef]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using β-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- Southern, C.; Cook, J.M.; Neetoo-Isseljee, Z.; Taylor, D.L.; Kettleborough, C.A.; Merritt, A.; Bassoni, D.L.; Raab, W.J.; Quinn, E.; Wehrman, T.S.; et al. Screening β-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J. Biomol. Screen. 2013, 18, 599–609. [Google Scholar] [CrossRef]
- Tutzauer, J.; Gonzalez de Valdivia, E.; Sward, K.; Alexandrakis Eilard, I.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. Ligand-independent G protein-coupled estrogen receptor/G protein-coupled receptor 30 activity: Lack of receptor-dependent effects of G-1 and 17β-estradiol. Mol. Pharmacol. 2021, 100, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Urban, N.; Leonhardt, M.; Schaefer, M. Multiplex G protein–coupled receptor screen reveals reliably acting agonists and a Gq-phospholipase C coupling mode of GPR30/GPER1. Mol. Pharmacol. 2023, 103, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Valdivia, E.; Sanden, C.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. Human G protein-coupled receptor 30 is N-glycosylated and N-terminal domain asparagine 44 is required for receptor structure and activity. Biosci. Rep. 2019, 39, BSR20182436. [Google Scholar] [CrossRef] [PubMed]
- Bomberger, J.M.; Spielman, W.S.; Hall, C.S.; Weinman, E.J.; Parameswaran, N. Receptor activity-modifying protein (RAMP) isoform-specific regulation of adrenomedullin receptor trafficking by NHERF-1. J. Biol. Chem. 2005, 280, 23926–23935. [Google Scholar] [CrossRef]
- Lenhart, P.M.; Broselid, S.; Barrick, C.J.; Leeb-Lundberg, L.M.; Caron, K.M. G-protein-coupled receptor 30 interacts with receptor activity-modifying protein 3 and confers sex-dependent cardioprotection. J. Mol. Endocrinol. 2013, 51, 191–202. [Google Scholar] [CrossRef]
- Broselid, S.; Berg, K.A.; Chavera, T.A.; Kahn, R.; Clarke, W.P.; Olde, B.; Leeb-Lundberg, L.M. G protein-coupled receptor 30 (GPR30) forms a plasma membrane complex with membrane-associated guanylate kinases (MAGUKs) and protein kinase A-anchoring protein 5 (AKAP5) that constitutively inhibits cAMP production. J. Biol. Chem. 2014, 289, 22117–22127. [Google Scholar] [CrossRef]
- Tran, Q.K.; VerMeer, M.; Burgard, M.A.; Hassan, A.B.; Giles, J. Hetero-oligomeric complex between the G protein-coupled estrogen receptor 1 and the plasma membrane Ca2+-ATPase 4b. J. Biol. Chem. 2015, 290, 13293–13307. [Google Scholar] [CrossRef]
- Paek, J.; Kalocsay, M.; Staus, D.P.; Wingler, L.; Pascolutti, R.; Paulo, J.A.; Gygi, S.P.; Kruse, A.C. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 2017, 169, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Hung, V.; Udeshi, N.D.; Lam, S.S.; Loh, K.H.; Cox, K.J.; Pedram, K.; Carr, S.A.; Ting, A.Y. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein-protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef] [PubMed]
- Gitter, A.; Klein-Seetharaman, J.; Gupta, A.; Bar-Joseph, Z. Discovering pathways by orienting edges in protein interaction networks. Nucleic Acids Res. 2011, 39, e22. [Google Scholar] [CrossRef]
- Navlakha, S.; Gitter, A.; Bar-Joseph, Z. A network-based approach for predicting missing pathway interactions. PLoS Comput. Biol. 2012, 8, e1002640. [Google Scholar] [CrossRef]
- Albanito, L.; Sisci, D.; Aquila, S.; Brunelli, E.; Vivacqua, A.; Madeo, A.; Lappano, R.; Pandey, D.P.; Picard, D.; Mauro, L.; et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology 2008, 149, 3799–3808. [Google Scholar] [CrossRef]
- Kaewsapsak, P.; Shechner, D.M.; Mallard, W.; Rinn, J.L.; Ting, A.Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 2017, 6, e29224. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Abrami, L.; Wang, W.-A.; Henry, C.; Frieden, M.; Didier, M.; van der Goot, F.G.; Demaurex, N. S-acylation by ZDHHC20 targets ORAI1 channels to lipid rafts for efficient Ca2+ signaling by Jurkat T cell receptors at the immune synapse. eLife 2021, 10, e72051. [Google Scholar] [CrossRef]
- Ge, Y.; Kang, Y.; Cassidy, R.M.; Moon, K.-M.; Lewis, R.; Wong, R.O.; Foster, L.J.; Craig, A.M. Clptm1 limits forward trafficking of GABAA receptors to scale inhibitory synaptic strength. Neuron 2018, 97, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.C.; Moon, S.U.; Kang, H.S.; Choi, H.S.; Han, H.D.; Kim, K.H. PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway. Nat. Commun. 2019, 10, 3185. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. 1D and 2D annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012, 13 (Suppl. S16), S12. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online parallel accumulation-serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Smith, A.; Cronan, J.E., Jr. Molecular biology of biotin attachment to proteins. J. Nutr. 1999, 129, 477S–484S. [Google Scholar] [CrossRef]
- Myers, S.A.; Wright, J.; Peckner, R.; Kalish, B.T.; Zhang, F.; Carr, S.A. Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat. Methods 2018, 15, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.; Xu, S.; Lv, W.; Shi, F.; Shan, A. IKBIP is a novel EMT-related biomarker and predicts poor survival in glioma. Transl. Neurosci. 2021, 12, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Chen, Z.; Zhao, L.; Ran, W.; Fan, J.; Hong, S.; Yang, Z. LAMB1 is related to the T stage and indicates poor prognosis in gastric cancer. Technol. Cancer Res. Treat. 2021, 20, 15330338211004944. [Google Scholar] [CrossRef]
- Pupo, M.; Bodmer, A.; Berto, M.; Maggiolini, M.; Dietrich, P.Y.; Picard, D. A genetic polymorphism repurposes the G-protein coupled and membrane-associated estrogen receptor GPER to a transcription factor-like molecule promoting paracrine signaling between stroma and breast carcinoma cells. Oncotarget 2017, 8, 46728–46744. [Google Scholar] [CrossRef]
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical AAA+ ATPases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549. [Google Scholar] [CrossRef]
- Hogan, P.G. The STIM1-ORAI1 microdomain. Cell Calcium 2015, 58, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.; Hawkins, A.; Zhang, Z.-S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Toshimori, T.; Noda, M.; Uchiyama, S.; Kato, K. Interaction mode between catalytic and regulatory subunits in glucosidase II involved in ER glycoprotein quality control. Protein Sci. 2016, 25, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kweon, Y.C.; Lee, A.R.; Lee, Y.Y.; Park, C.Y. Metastasis enhancer PGRMC1 boosts store-operated Ca2+ entry by uncoiling Ca2+ sensor STIM1 for focal adhesion turnover and actomyosin formation. Cell Rep. 2022, 38, 110281. [Google Scholar] [CrossRef]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef]
- Niedernberg, A.; Blaukat, A.; Schöneberg, T.; Kostenis, E. Regulated and constitutive activation of specific signalling pathways by the human S1P5 receptor. Br. J. Pharmacol. 2003, 138, 481–493. [Google Scholar] [CrossRef]
- Madeo, A.; Maggiolini, M. Nuclear alternate estrogen receptor GPR30 mediates 17β-estradiol–induced gene expression and migration in breast cancer–associated fibroblasts. Cancer Res. 2010, 70, 6036–6046. [Google Scholar] [CrossRef]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807. [Google Scholar] [CrossRef]
- Gobeil, F.; Fortier, A.; Zhu, T.; Bossolasco, M.; Leduc, M.; Grandbois, M.; Heveker, N.; Bkaily, G.; Chemtob, S.; Barbaz, D. G-protein-coupled receptors signalling at the cell nucleus: An emerging paradigm. Can. J. Physiol. Pharmacol. 2006, 84, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Jong, Y.-J.I.; Harmon, S.K.; Kumar, V.; O’Malley, K.L. Sequences within the C terminus of the metabotropic glutamate receptor 5 (mGluR5) are responsible for inner nuclear membrane localization. J. Biol. Chem. 2017, 292, 3637–3655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Zhou, J.Y.; Zhang, P.; Lin, L.; Mei, R.; Zhang, F.L.; Chen, Y.M.; Li, R. Clptm1, a new target in suppressing epileptic seizure by regulating GABAAR-mediated inhibitory synaptic transmission in a PTZ-induced epilepsy model. Kaohsiung J. Med. Sci. 2023, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nairn, A.V.; Moremen, K.W. Glucosidase, Alpha Neutral AB; Glucosidase II Subunit Beta (GANAB, PRKCSH, α-Glucosidase II). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 1283–1295. [Google Scholar]
- Gao, H.; Wang, Y.; Wegierski, T.; Skouloudaki, K.; Pütz, M.; Fu, X.; Engel, C.; Boehlke, C.; Peng, H.; Kuehn, E.W. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum. Mol. Genet. 2010, 19, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Forough, R.; Lindner, L.; Partridge, C.; Jones, B.; Guy, G.; Clark, G. Elevated 80K-H protein in breast cancer: A role for FGF-1 stimulation of 80K-H. Int. J. Biol. Markers 2003, 18, 89–98. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Do Heo, W.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 2010, 21, 1897–1907. [Google Scholar] [CrossRef]
- Hirve, N.; Rajanikanth, V.; Hogan, P.G.; Gudlur, A. Coiled-coil formation conveys a STIM1 signal from ER lumen to cytoplasm. Cell Rep. 2018, 22, 72–83. [Google Scholar] [CrossRef]
- Sohn, P.; McLaughlin, M.R.; Krishnan, P.; Wu, W.; Rupnik, M.S.; Takasu, A.; Senda, T.; Lee, C.-C.; Kono, T.; Evans-Molina, C. Stromal Interaction molecule 1 maintains β cell identity and function in female mice through preservation of G protein-coupled estrogen receptor 1 signaling. Diabetes 2023, 72, 1433–1445. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmadian Elmi, M.; Motamed, N.; Picard, D. Proteomic Analyses of the G Protein-Coupled Estrogen Receptor GPER1 Reveal Constitutive Links to Endoplasmic Reticulum, Glycosylation, Trafficking, and Calcium Signaling. Cells 2023, 12, 2571. https://doi.org/10.3390/cells12212571
Ahmadian Elmi M, Motamed N, Picard D. Proteomic Analyses of the G Protein-Coupled Estrogen Receptor GPER1 Reveal Constitutive Links to Endoplasmic Reticulum, Glycosylation, Trafficking, and Calcium Signaling. Cells. 2023; 12(21):2571. https://doi.org/10.3390/cells12212571
Chicago/Turabian StyleAhmadian Elmi, Maryam, Nasrin Motamed, and Didier Picard. 2023. "Proteomic Analyses of the G Protein-Coupled Estrogen Receptor GPER1 Reveal Constitutive Links to Endoplasmic Reticulum, Glycosylation, Trafficking, and Calcium Signaling" Cells 12, no. 21: 2571. https://doi.org/10.3390/cells12212571