
Mechanisms Underlying Rare Inherited Pediatric Retinal Vascular Diseases: FEVR, Norrie Disease, Persistent Fetal Vascular Syndrome


Abstract
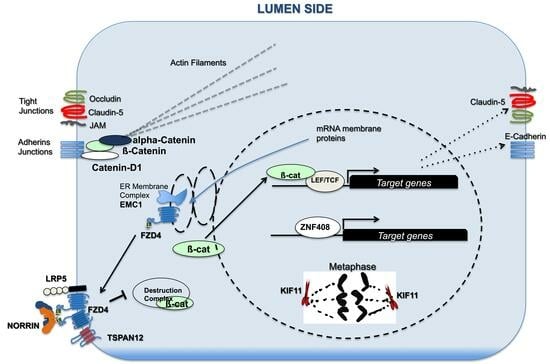
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genes and Proteins
2.1. KIF11
2.2. ZNF408
2.3. CTNNB1
2.4. NDP
2.5. FZD4
2.6. LRP5
2.7. TSPAN-12
2.8. Notable Recent Genes Linked to FEVR Phenotypes
2.9. EMC-1
2.10. CTNNA1
2.11. CTNND1
3. Conclusions
- As we survey the protein domain maps that we have presented of currently known pathogenic and likely pathogenic variants, we can conclude that almost any functional subdomain of these proteins can be involved. This is not surprising because important protein-interaction functions or structural functions exist throughout their entire amino acid sequences. There are few, if any, non-essential regions in the ten proteins reviewed. This would suggest that there may be many novel variants awaiting discovery in the human population, especially for more recently linked genes such as CTNNA1;
- FEVR can involve the disruption of any one of several different functions in endothelial cells, not just those related directly to the Norrin Wnt-signaling pathway. This is established by disease-causing variants in ZNF408, KIF11, and EMC1. However, what all these genes and their protein products have in common is that they are particularly important for critical retinal endothelial cell functions. These include correct membrane insertion of transmembrane proteins in the ER, regulation of adherens and tight junctions, cell growth, cell proliferation, migration of endothelial cells during formation of the retinal vasculature, and maturation of a high-barrier character endothelium. It is possible, and expected, that we may continue to discover novel FEVR-linked genes and good candidates would be any gene that is particularly enriched in retinal endothelial cells versus other endothelial cells. However, more broadly expressed genes with roles in any of the above noted functions may be good candidates as well. That may include nine other genes for subunits of the Endoplasmic Reticulum Membrane Complex;
- The multiple allele knockout studies by Junge et al. (2009) in mice suggested the possibility that more severe FEVR-like phenotypes could result from combinations of two or more different alleles that have a minimal impact alone [66]. Our group recently surveyed a cohort of FEVR patients and immediate relatives to confirm that the incidence of protein-altering variants in two or three different FEVR-linked genes was substantially greater than in the general population [17]. Thus, it is possible that combined mild-alleles might result in more severe phenotypes, but we have not yet described clear examples of this in FEVR. To explore such possibilities for this relatively rare condition, it will be helpful to apply genetic testing methods that survey as many genes as possible and to form global research collaborations to investigate genotypes and phenotypes from varied populations.
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Fruttiger, M. Development of the Mouse Retinal Vasculature: Angiogenesis versus Vasculogenesis. Investig. Ophthalmol. Vis. Sci. 2002, 43, 522–527. [Google Scholar]
- Zuercher, J.; Fritzsche, M.; Feil, S.; Mohn, L.; Berger, W. Norrin Stimulates Cell Proliferation in the Superficial Retinal Vascular Plexus and Is Pivotal for the Recruitment of Mural Cells. Hum. Mol. Genet. 2012, 21, 2619–2630. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal Vasculature Development in Health and Disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Tokunaga, C.C.; Mitton, K.P.; Dailey, W.; Massoll, C.; Roumayah, K.; Guzman, E.; Tarabishy, N.; Cheng, M.; Drenser, K.A. Effects of Anti-VEGF Treatment on the Recovery of the Developing Retina Following Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Dailey, W.A.; Drenser, K.A.; Wong, S.C.; Cheng, M.; Vercellone, J.; Roumayah, K.K.; Feeney, E.V.; Deshpande, M.; Guzman, A.E.; Trese, M.; et al. Ocular Coherence Tomography Image Data of the Retinal Laminar Structure in a Mouse Model of Oxygen-Induced Retinopathy. Data Brief 2017, 15, 491–495. [Google Scholar] [CrossRef]
- Dailey, W.A.; Drenser, K.A.; Wong, S.C.; Cheng, M.; Vercellone, J.; Roumayah, K.K.; Feeney, E.V.; Deshpande, M.; Guzman, A.E.; Trese, M.; et al. Norrin Treatment Improves Ganglion Cell Survival in an Oxygen-Induced Retinopathy Model of Retinal Ischemia. Exp. Eye Res. 2017, 164, 129–138. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood–Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef]
- Glenney, J.R.; Soppet, D. Sequence and Expression of Caveolin, a Protein Component of Caveolae Plasma Membrane Domains Phosphorylated on Tyrosine in Rous Sarcoma Virus-Transformed Fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10517–10521. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Antonetti, D.A.; Barber, A.J.; Lanoue, K.F.; Levison, S.W.; Penn State Retina Research Group. Diabetic Retinopathy: More Than Meets the Eye. Surv. Ophthalmol. 2002, 47, S253–S262. [Google Scholar] [CrossRef]
- Feng, Y.; Venema, V.J.; Venema, R.C.; Tsai, N.; Caldwell, R.B. VEGF Induces Nuclear Translocation of Flk-1/KDR, Endothelial Nitric Oxide Synthase, and Caveolin-1 in Vascular Endothelial Cells. Biochem. Biophys. Res. Commun. 1999, 256, 192–197. [Google Scholar] [CrossRef]
- Wu, W.-C.; Drenser, K.; Trese, M.; Capone, A., Jr.; Dailey, W. Retinal Phenotype-Genotype Correlation of Pediatric Patients Expressing Mutations in the Norrie Disease Gene. Arch. Ophthalmol. 2007, 125, 225–230. [Google Scholar] [CrossRef]
- Drenser, K.A.; Trese, M.T. Familial Exudative Vitreoretinopathy and Osteoporosis-Pseudoglioma Syndrome Caused by a Mutation in the LRP5 Gene. Arch. Ophthalmol. 2007, 125, 431–432. [Google Scholar] [CrossRef]
- Drenser, K.A.; Dailey, W.; Capone, A.; Trese, M.T. Genetic Evaluation to Establish the Diagnosis of X-Linked Familial Exudative Vitreoretinopathy. Ophthalmic Genet. 2006, 27, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Criswick, V.G.; Schepens, C.L. Familial Exudative Vitreoretinopathy. Am. J. Ophthalmol. 1969, 68, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Ranchod, T.M.; Ho, L.Y.; Drenser, K.A.; Capone, A.; Trese, M.T. Clinical Presentation of Familial Exudative Vitreoretinopathy. Ophthalmology 2011, 118, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Scruggs, B.A.; Reding, M.Q.; Schimmenti, L.A. NDP-Related Retinopathies. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Cicerone, A.P.; Dailey, W.; Sun, M.; Santos, A.; Jeong, D.; Jones, L.; Koustas, K.; Drekh, M.; Schmitz, K.; Haque, N.; et al. A Survey of Multigenic Protein-Altering Variant Frequency in Familial Exudative Vitreo-Retinopathy (FEVR) Patients by Targeted Sequencing of Seven FEVR-Linked Genes. Genes 2022, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.W.; Stem, M.S.; Schuette, J.L.; Keegan, C.E.; Besirli, C.G. CTNNB1 Mutation Associated with Familial Exudative Vitreoretinopathy (FEVR) Phenotype. Ophthalmic Genet. 2016, 37, 468–470. [Google Scholar] [CrossRef]
- Li, J.K.; Fei, P.; Li, Y.; Huang, Q.J.; Zhang, Q.; Zhang, X.; Rao, Y.Q.; Li, J.; Zhao, P. Identification of Novel KIF11 Mutations in Patients with Familial Exudative Vitreoretinopathy and a Phenotypic Analysis. Sci. Rep. 2016, 6, 26564. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Nikopoulos, K.; Dona, M.; Gilissen, C.; Hoischen, A.; Boonstra, F.N.; Poulter, J.A.; Kondo, H.; Berger, W.; Toomes, C.; et al. ZNF408 Is Mutated in Familial Exudative Vitreoretinopathy and Is Crucial for the Development of Zebrafish Retinal Vasculature. Proc. Natl. Acad. Sci. USA 2013, 110, 9856–9861. [Google Scholar] [CrossRef]
- Yang, M.; Li, S.; Huang, L.; Zhao, R.; Dai, E.; Jiang, X.; He, Y.; Lu, J.; Peng, L.; Liu, W.; et al. CTNND1 Variants Cause Familial Exudative Vitreoretinopathy through the Wnt/Cadherin Axis. JCI Insight 2022, 7, e158428. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, M.; Zhao, P.; Li, S.; Zhang, L.; Huang, L.; Huang, Y.; Fei, P.; Yang, Y.; Zhang, S.; et al. Catenin α 1 Mutations Cause Familial Exudative Vitreoretinopathy by Overactivating Norrin/β-Catenin Signaling. J. Clin. Investig. 2021, 131, e139869. [Google Scholar] [CrossRef]
- Li, S.; Yang, M.; Zhao, R.; Peng, L.; Liu, W.; Jiang, X.; He, Y.; Dai, E.; Zhang, L.; Yang, Y.; et al. Defective EMC1 Drives Abnormal Retinal Angiogenesis via Wnt/β-Catenin Signaling and May Be Associated with the Pathogenesis of Familial Exudative Vitreoretinopathy. Genes Dis. 2023, 10, 2572–2585. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Kashina, A.S.; Baskin, R.J.; Cole, D.G.; Wedaman, K.P.; Saxton, W.M.; Scholey, J.M. A Bipolar Kinesin. Nature 1996, 379, 270–272. [Google Scholar] [CrossRef]
- Jones, G.E.; Ostergaard, P.; Moore, A.T.; Connell, F.C.; Williams, D.; Quarrell, O.; Brady, A.F.; Spier, I.; Hazan, F.; Moldovan, O.; et al. Microcephaly with or without Chorioretinopathy, Lymphoedema, or Mental Retardation (MCLMR): Review of Phenotype Associated with KIF11 Mutations. Eur. J. Hum. Genet. 2014, 22, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Mirzaa, G.M.; Enyedi, L.; Parsons, G.; Collins, S.; Medne, L.; Adams, C.; Ward, T.; Davitt, B.; Bicknese, A.; Zackai, E.; et al. Congenital Microcephaly and Chorioretinopathy Due to de Novo Heterozygous KIF11 Mutations: Five Novel Mutations and Review of the Literature. Am. J. Med. Genet. A 2014, 164, 2879–2886. [Google Scholar] [CrossRef]
- Wang, Y.; Smallwood, P.M.; Williams, J.; Nathans, J. A Mouse Model for Kinesin Family Member 11 (Kif11)-Associated Familial Exudative Vitreoretinopathy. Hum. Mol. Genet. 2020, 29, 1121–1131. [Google Scholar] [CrossRef]
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology 2013, 2, 107–141. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-Finger Proteins in Health and Disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Avila-Fernandez, A.; Perez-Carro, R.; Corton, M.; Lopez-Molina, M.I.; Campello, L.; Garanto, A.; Fernandez-Sanchez, L.; Duijkers, L.; Lopez-Martinez, M.A.; Riveiro-Alvarez, R.; et al. Whole-Exome Sequencing Reveals ZNF408 as a New Gene Associated with Autosomal Recessive Retinitis Pigmentosa with Vitreal Alterations. Hum. Mol. Genet. 2015, 24, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Karjosukarso, D.W.; van Gestel, S.H.C.; Qu, J.; Kouwenhoven, E.N.; Duijkers, L.; Garanto, A.; Zhou, H.; Collin, R.W.J. An FEVR-Associated Mutation in ZNF408 Alters the Expression of Genes Involved in the Development of Vasculature. Hum. Mol. Genet. 2018, 27, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kimelman, D. Mechanistic Insights from Structural Studies of Beta-Catenin and Its Binding Partners. J. Cell Sci. 2007, 120, 3337–3344. [Google Scholar] [CrossRef]
- Huber, A.H.; Weis, W.I. The Structure of the Beta-Catenin/E-Cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by Beta-Catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Taylor, R.L.; Soriano, C.S.; Williams, S.; Dzulova, D.; Ashworth, J.; Hall, G.; Gale, T.; Lloyd, I.C.; Inglehearn, C.F.; Toomes, C.; et al. Bi-Allelic Mutation of CTNNB1 Causes a Severe Form of Syndromic Microphthalmia, Persistent Foetal Vasculature and Vitreoretinal Dysplasia. Orphanet J. Rare Dis. 2022, 17, 110. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, A.; Tamm, E.R. Norrin: Molecular and Functional Properties of an Angiogenic and Neuroprotective Growth Factor. Prog. Retin. Eye Res. 2012, 31, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Zeilbeck, L.F.; Müller, B.B.; Leopold, S.A.; Senturk, B.; Langmann, T.; Tamm, E.R.; Ohlmann, A. Norrin Mediates Angiogenic Properties via the Induction of Insulin-like Growth Factor-1. Exp. Eye Res. 2016, 145, 317–326. [Google Scholar] [CrossRef]
- Shastry, B.S.; Hejtmancik, J.F.; Trese, M.T. Identification of Novel Missense Mutations in the Norrie Disease Gene Associated with One X-Linked and Four Sporadic Cases of Familial Exudative Vitreoretinopathy. Hum. Mutat. 1997, 9, 396–401. [Google Scholar] [CrossRef]
- Ke, J.; Harikumar, K.G.; Erice, C.; Chen, C.; Gu, X.; Wang, L.; Parker, N.; Cheng, Z.; Xu, W.; Williams, B.O.; et al. Structure and Function of Norrin in Assembly and Activation of a Frizzled 4-Lrp5/6 Complex. Genes Dev. 2013, 27, 2305–2319. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular Development in the Retina and Inner Ear: Control by Norrin and Frizzled-4, a High-Affinity Ligand-Receptor Pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef]
- Xiao, H.; Tong, Y.; Zhu, Y.; Peng, M. Familial Exudative Vitreoretinopathy-Related Disease-Causing Genes and Norrin/β-Catenin Signal Pathway: Structure, Function, and Mutation Spectrums. J. Ophthalmol. 2019, 2019, 5782536. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, C.; Williams, J.; Smallwood, P.M.; Zhang, C.; Junge, H.J.; Nathans, J. Interplay of the Norrin and Wnt7a/Wnt7b Signaling Systems in Blood-Brain Barrier and Blood-Retina Barrier Development and Maintenance. Proc. Natl. Acad. Sci. USA 2018, 115, E11827–E11836. [Google Scholar] [CrossRef]
- Ye, X.; Wang, Y.; Nathans, J. The Norrin/Frizzled4 Signaling Pathway in Retinal Vascular Development and Disease. Trends Mol. Med. 2010, 16, 417–425. [Google Scholar] [CrossRef]
- Ohlmann, A.; Scholz, M.; Goldwich, A.; Chauhan, B.K.; Hudl, K.; Ohlmann, A.V.; Zrenner, E.; Berger, W.; Cvekl, A.; Seeliger, M.W.; et al. Ectopic Norrin Induces Growth of Ocular Capillaries and Restores Normal Retinal Angiogenesis in Norrie Disease Mutant Mice. J. Neurosci. 2005, 25, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, C.C.; Chen, Y.-H.; Dailey, W.; Cheng, M.; Drenser, K.A. Retinal Vascular Rescue of Oxygen-Induced Retinopathy in Mice by Norrin. Investig. Opthalmol. Vis. Sci. 2013, 54, 222. [Google Scholar] [CrossRef]
- Pauzuolyte, V.; Patel, A.; Wawrzynski, J.R.; Ingham, N.J.; Leong, Y.C.; Karda, R.; Bitner-Glindzicz, M.; Berger, W.; Waddington, S.N.; Steel, K.P.; et al. Systemic Gene Therapy Rescues Retinal Dysfunction and Hearing Loss in a Model of Norrie Disease. EMBO Mol Med 2023, 15, e17393. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Lin, C.-M.; Liebner, S.; Antonetti, D.A. Norrin Restores Blood-Retinal Barrier Properties after Vascular Endothelial Growth Factor-Induced Permeability. J. Biol. Chem. 2020, 295, 4647–4660. [Google Scholar] [CrossRef] [PubMed]
- Bang, I.; Kim, H.R.; Beaven, A.H.; Kim, J.; Ko, S.B.; Lee, G.R.; Lee, H.; Im, W.; Seok, C.; Chung, K.Y.; et al. Biophysical and Functional Characterization of Norrin Signaling through Frizzled4. Proc. Natl. Acad. Sci. USA 2018, 115, 8787–8792. [Google Scholar] [CrossRef]
- Paes, K.T.; Wang, E.; Henze, K.; Vogel, P.; Read, R.; Suwanichkul, A.; Kirkpatrick, L.L.; Potter, D.; Newhouse, M.M.; Rice, D.S. Frizzled 4 Is Required for Retinal Angiogenesis and Maintenance of the Blood-Retina Barrier. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6452–6461. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Hayashi, H.; Oshima, K.; Tahira, T.; Hayashi, K. Frizzled 4 Gene (FZD4) Mutations in Patients with Familial Exudative Vitreoretinopathy with Variable Expressivity. Br. J. Ophthalmol. 2003, 87, 1291–1295. [Google Scholar] [CrossRef]
- Dailey, W.A.; Gryc, W.; Garg, P.G.; Drenser, K.A. Frizzled-4 Variations Associated with Retinopathy and Intrauterine Growth Retardation. Ophthalmology 2015, 122, 1917–1923. [Google Scholar] [CrossRef]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL Receptor-Related Proteins 5 and 6 in Wnt/Beta-Catenin Signaling: Arrows Point the Way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Yablonka-Reuveni, Z.; Gong, X. LRP5 Is Required for Vascular Development in Deeper Layers of the Retina. PLoS ONE 2010, 5, e11676. [Google Scholar] [CrossRef]
- Huang, W.; Li, Q.; Amiry-Moghaddam, M.; Hokama, M.; Sardi, S.H.; Nagao, M.; Warman, M.L.; Olsen, B.R. Critical Endothelial Regulation by LRP5 during Retinal Vascular Development. PLoS ONE 2016, 11, e0152833. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Hayashi, H.; Oshima, K.; Tahira, T.; Hayashi, K.; Kondo, H. Complexity of the Genotype-Phenotype Correlation in Familial Exudative Vitreoretinopathy with Mutations Im the LRP5 and/or FZD4 Genes. Hum. Mutat. 2005, 26, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Seigneuret, M.; Delaguillaumie, A.; Lagaudrière-Gesbert, C.; Conjeaud, H. Structure of the Tetraspanin Main Extracellular Domain: A Partially Conserved Fold with a Structurally Variable Domain Insertion. J. Biol. Chem. 2001, 276, 40055–40064. [Google Scholar] [CrossRef]
- Poulter, J.A.; Ali, M.; Gilmour, D.F.; Rice, A.; Kondo, H.; Hayashi, K.; Mackey, D.A.; Kearns, L.S.; Ruddle, J.B.; Craig, J.E.; et al. Mutations in TSPAN12 Cause Autosomal-Dominant Familial Exudative Vitreoretinopathy. Am. J. Hum. Genet. 2010, 86, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a Glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Mohd Khair, S.Z.N.; Ismail, A.S.; Embong, Z.; Mohamed Yusoff, A.A. Detection of FZD4, LRP5 and TSPAN12 Genes Variants in Malay Premature Babies with Retinopathy of Prematurity. J. Ophthalmic Vis. Res. 2019, 14, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lai, M.B.; Pedler, M.G.; Johnson, V.; Adams, R.H.; Petrash, J.M.; Chen, Z.; Junge, H.J. Endothelial Cell–Specific Inactivation of TSPAN12 (Tetraspanin 12) Reveals Pathological Consequences of Barrier Defects in an Otherwise Intact Vasculature. Arter. Thromb. Vasc. Biol. 2018, 38, 2691–2705. [Google Scholar] [CrossRef] [PubMed]
- Nikopoulos, K.; Gilissen, C.; Hoischen, A.; Erik van Nouhuys, C.; Boonstra, F.N.; Blokland, E.A.W.; Arts, P.; Wieskamp, N.; Strom, T.M.; Ayuso, C.; et al. Next-Generation Sequencing of a 40 Mb Linkage Interval Reveals TSPAN12 Mutations in Patients with Familial Exudative Vitreoretinopathy. Am. J. Hum. Genet. 2010, 86, 240–247. [Google Scholar] [CrossRef]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 Regulates Retinal Vascular Development by Promoting Norrin- but Not Wnt-Induced FZD4/β-Catenin Signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Harel, T.; Yesil, G.; Bayram, Y.; Coban-Akdemir, Z.; Charng, W.L.; Karaca, E.; Al Asmari, A.; Eldomery, M.K.; Hunter, J.V.; Jhangiani, S.N.; et al. Monoallelic and Biallelic Variants in EMC1 Identified in Individuals with Global Developmental Delay, Hypotonia, Scoliosis, and Cerebellar Atrophy. Am. J. Hum. Genet. 2016, 98, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Hong, S.; Indra, I.; Sergeeva, A.P.; Troyanovsky, R.B.; Shapiro, L.; Honig, B.; Troyanovsky, S.M. α-Catenin-Mediated Cadherin Clustering Couples Cadherin and Actin Dynamics. J. Cell Biol. 2015, 210, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Majewski, I.J.; Kluijt, I.; Cats, A.; Scerri, T.S.; de Jong, D.; Kluin, R.J.C.; Hansford, S.; Hogervorst, F.B.L.; Bosma, A.J.; Hofland, I.; et al. An α-E-Catenin (CTNNA1) Mutation in Hereditary Diffuse Gastric Cancer. J. Pathol. 2013, 229, 621–629. [Google Scholar] [CrossRef]
- Piao, H.-L.; Yuan, Y.; Wang, M.; Sun, Y.; Liang, H.; Ma, L. α-Catenin Acts as a Tumour Suppressor in E-Cadherin-Negative Basal-like Breast Cancer by Inhibiting NF-ΚB Signalling. Nat. Cell Biol. 2014, 16, 245–254. [Google Scholar] [CrossRef]
- Saksens, N.T.M.; Krebs, M.P.; Schoenmaker-Koller, F.E.; Hicks, W.; Yu, M.; Shi, L.; Rowe, L.; Collin, G.B.; Charette, J.R.; Letteboer, S.J.; et al. Mutations in CTNNA1 Cause Butterfly-Shaped Pigment Dystrophy and Perturbed Retinal Pigment Epithelium Integrity. Nat. Genet. 2016, 48, 144–151. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Alharatani, R.; Ververi, A.; Beleza-Meireles, A.; Ji, W.; Mis, E.; Patterson, Q.T.; Griffin, J.N.; Bhujel, N.; Chang, C.A.; Dixit, A.; et al. Novel Truncating Mutations in CTNND1 Cause a Dominant Craniofacial and Cardiac Syndrome. Hum. Mol. Genet. 2020, 29, 1900. [Google Scholar] [CrossRef]
- Ishiyama, N.; Lee, S.H.; Liu, S.; Li, G.Y.; Smith, M.J.; Reichardt, L.F.; Ikura, M. Dynamic and Static Interactions between P120 Catenin and E-Cadherin Regulate the Stability of Cell-Cell Adhesion. Cell 2010, 141, 117–128. [Google Scholar] [CrossRef]
- Anastasiadis, P.Z.; Moon, S.Y.; Thoreson, M.A.; Mariner, D.J.; Crawford, H.C.; Zheng, Y.; Reynolds, A.B. Inhibition of RhoA by P120 Catenin. Nat. Cell Biol. 2000, 2, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Kim, S.W.; Lyons, J.P.; Ji, H.; Nguyen, T.T.; Cho, K.; Barton, M.C.; Deroo, T.; Vleminckx, K.; McCrea, P.D. Kaiso/P120-Catenin and TCF/β-Catenin Complexes Coordinately Regulate Canonical Wnt Gene Targets. Dev. Cell 2005, 8, 843–854. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, V.; Abdelmessih, G.; Dailey, W.A.; Pinnock, C.; Jobczyk, V.; Rashingkar, R.; Drenser, K.A.; Mitton, K.P. Mechanisms Underlying Rare Inherited Pediatric Retinal Vascular Diseases: FEVR, Norrie Disease, Persistent Fetal Vascular Syndrome. Cells 2023, 12, 2579. https://doi.org/10.3390/cells12212579
Le V, Abdelmessih G, Dailey WA, Pinnock C, Jobczyk V, Rashingkar R, Drenser KA, Mitton KP. Mechanisms Underlying Rare Inherited Pediatric Retinal Vascular Diseases: FEVR, Norrie Disease, Persistent Fetal Vascular Syndrome. Cells. 2023; 12(21):2579. https://doi.org/10.3390/cells12212579
Chicago/Turabian StyleLe, Vincent, Gabrielle Abdelmessih, Wendy A. Dailey, Cecille Pinnock, Victoria Jobczyk, Revati Rashingkar, Kimberly A. Drenser, and Kenneth P. Mitton. 2023. "Mechanisms Underlying Rare Inherited Pediatric Retinal Vascular Diseases: FEVR, Norrie Disease, Persistent Fetal Vascular Syndrome" Cells 12, no. 21: 2579. https://doi.org/10.3390/cells12212579
APA StyleLe, V., Abdelmessih, G., Dailey, W. A., Pinnock, C., Jobczyk, V., Rashingkar, R., Drenser, K. A., & Mitton, K. P. (2023). Mechanisms Underlying Rare Inherited Pediatric Retinal Vascular Diseases: FEVR, Norrie Disease, Persistent Fetal Vascular Syndrome. Cells, 12(21), 2579. https://doi.org/10.3390/cells12212579