

Ultrastructural Abnormalities in Induced Pluripotent Stem Cell-Derived Neural Stem Cells and Neurons of Two Cohen Syndrome Patients

 , , , , , , , and
, , , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Sample Collection
2.2. Primary Cell Lines and Culture Conditions
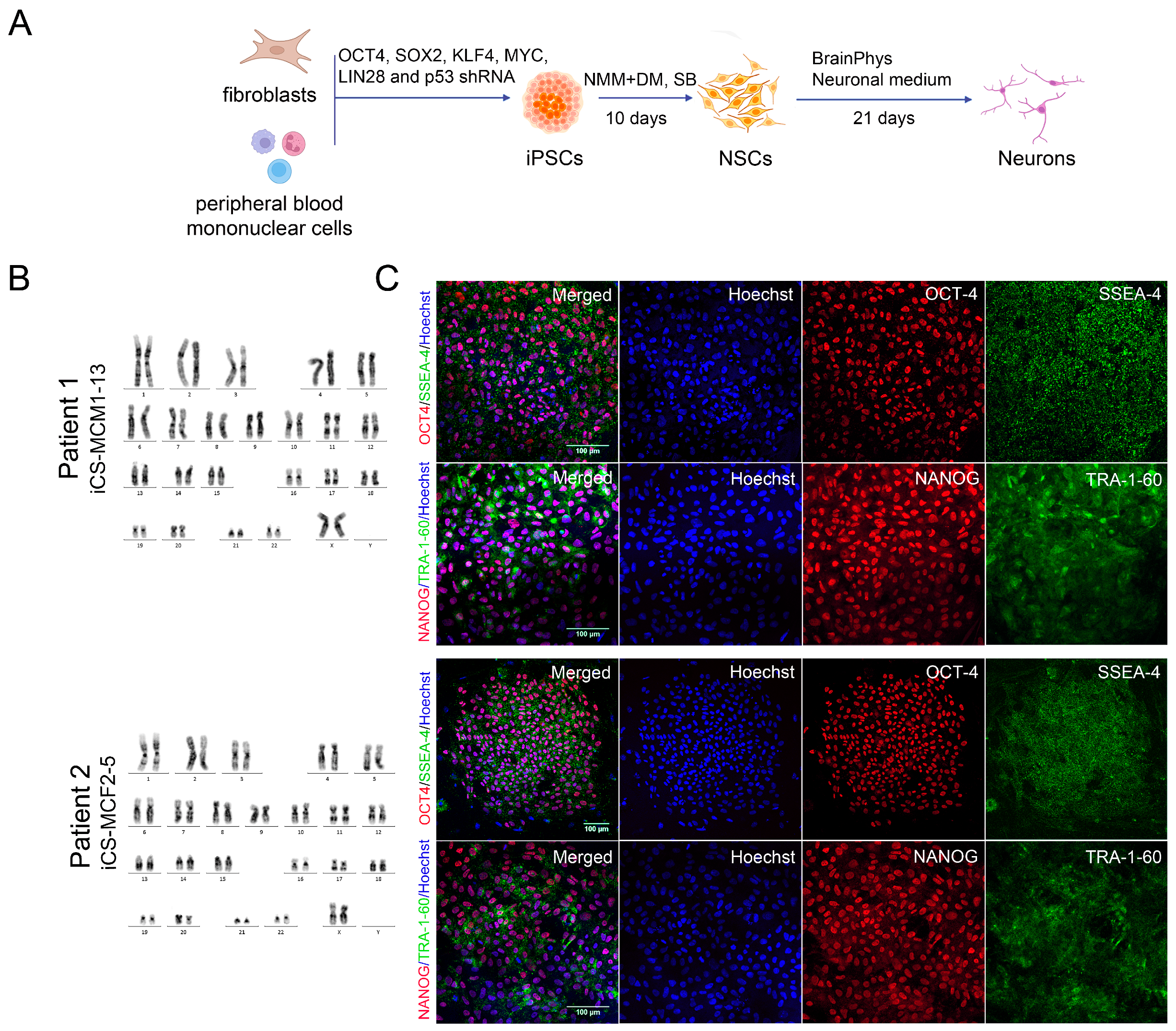
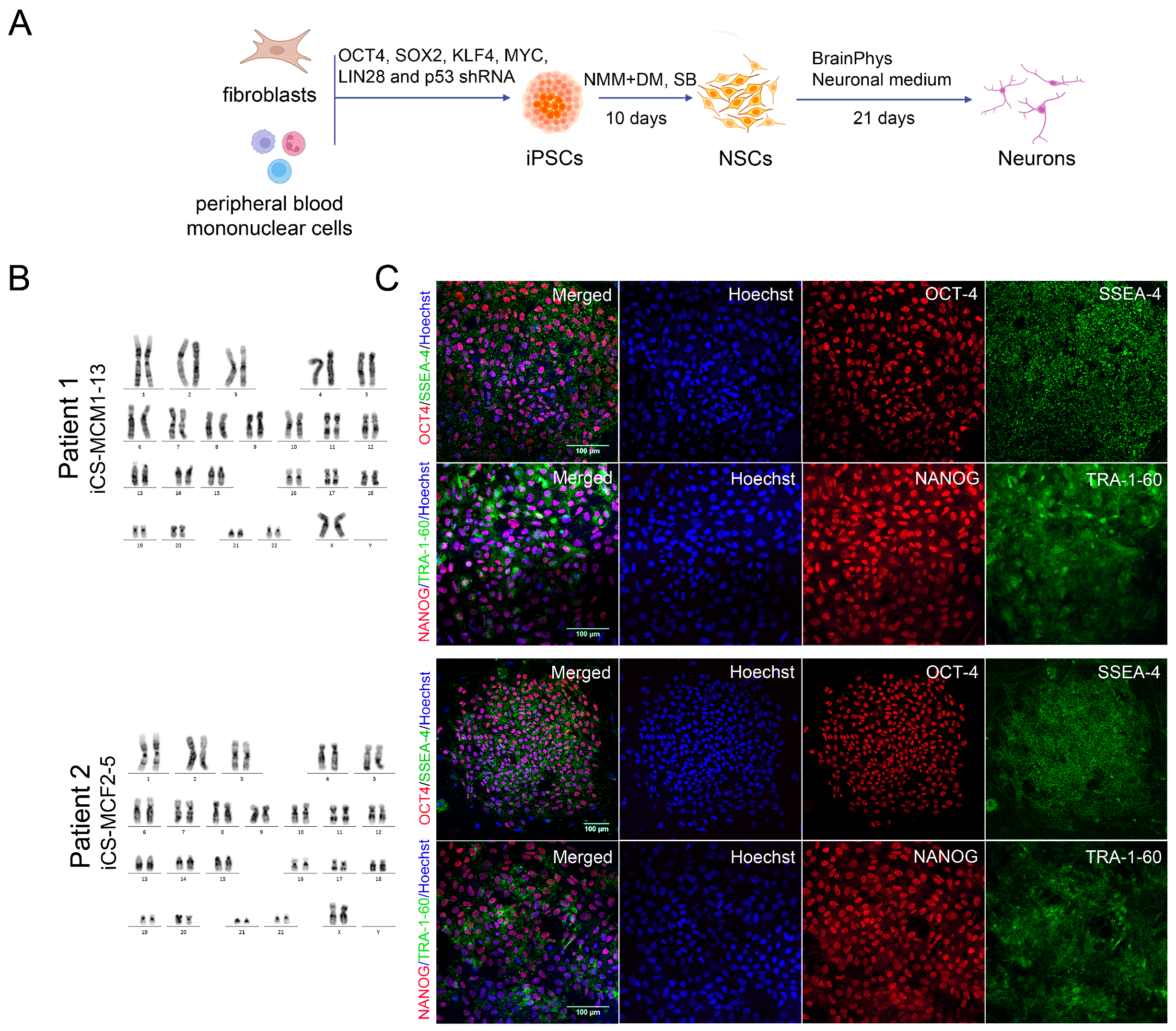
2.3. Generation of iPSCs
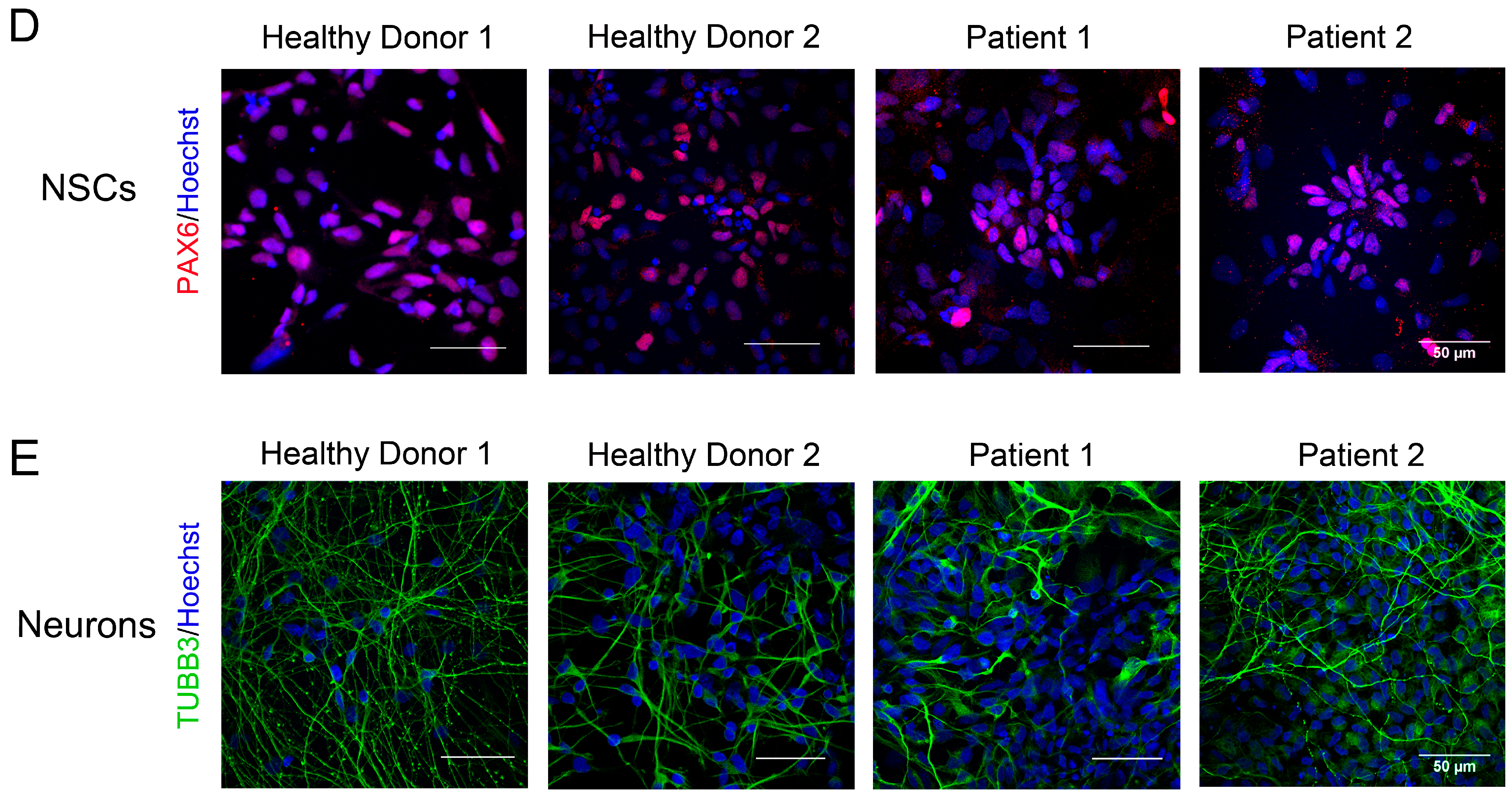
2.4. Neural Induction by Dual Inhibition of SMAD Signaling
2.5. Immunocytochemistry and Fluorescence Microscopy
2.6. Lysotracker Labeling
2.7. Transmission Electron Microscopy
2.8. RNA Isolation and Reverse Transcription
2.9. RT-PCR
2.10. Quantitative RT-PCR
2.11. DNA Isolation
2.12. Whole-Exome Sequencing and Sanger Sequencing Validation
2.13. Karyotype Analysis
2.14. Quantification and Statistical Analysis
3. Results
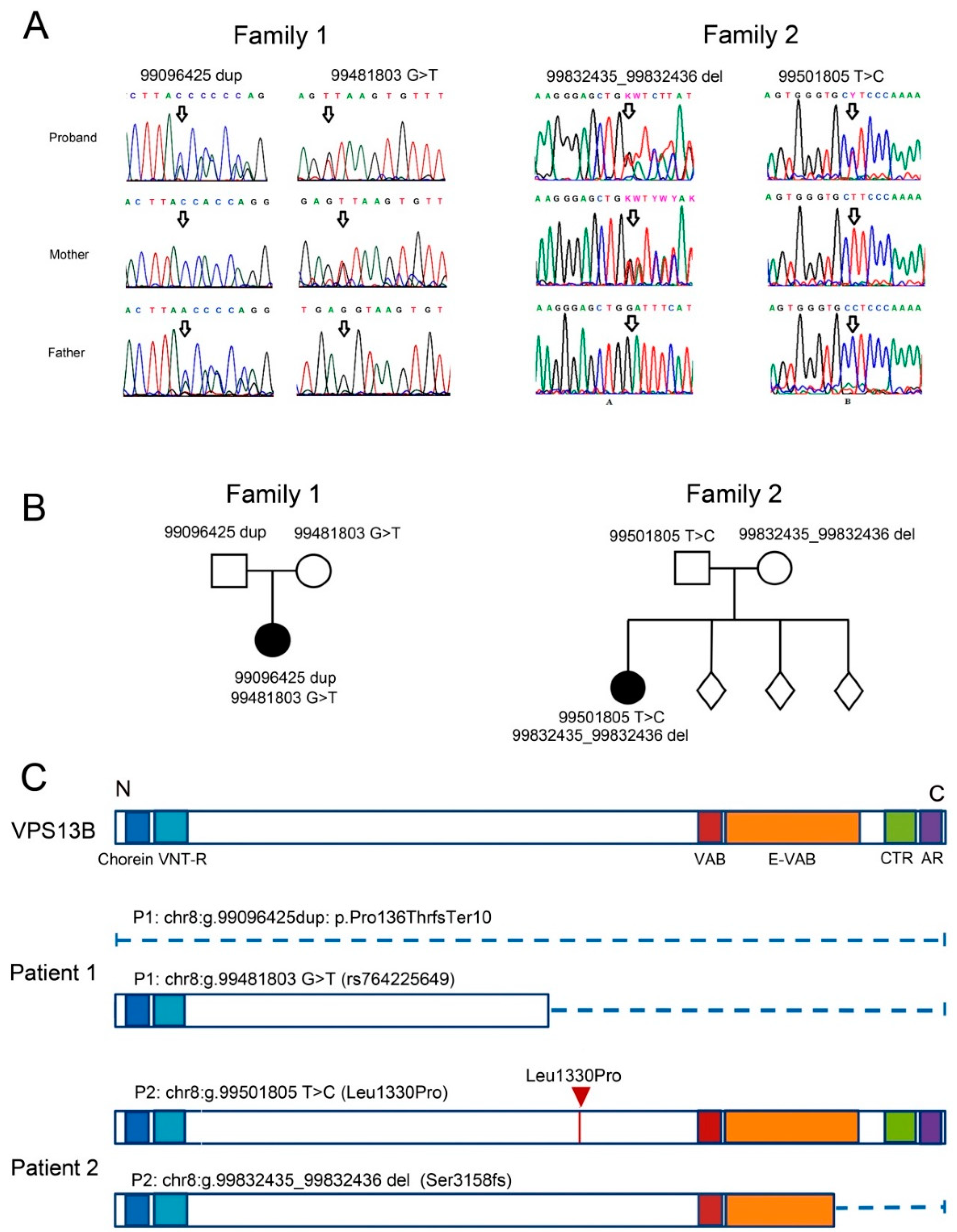
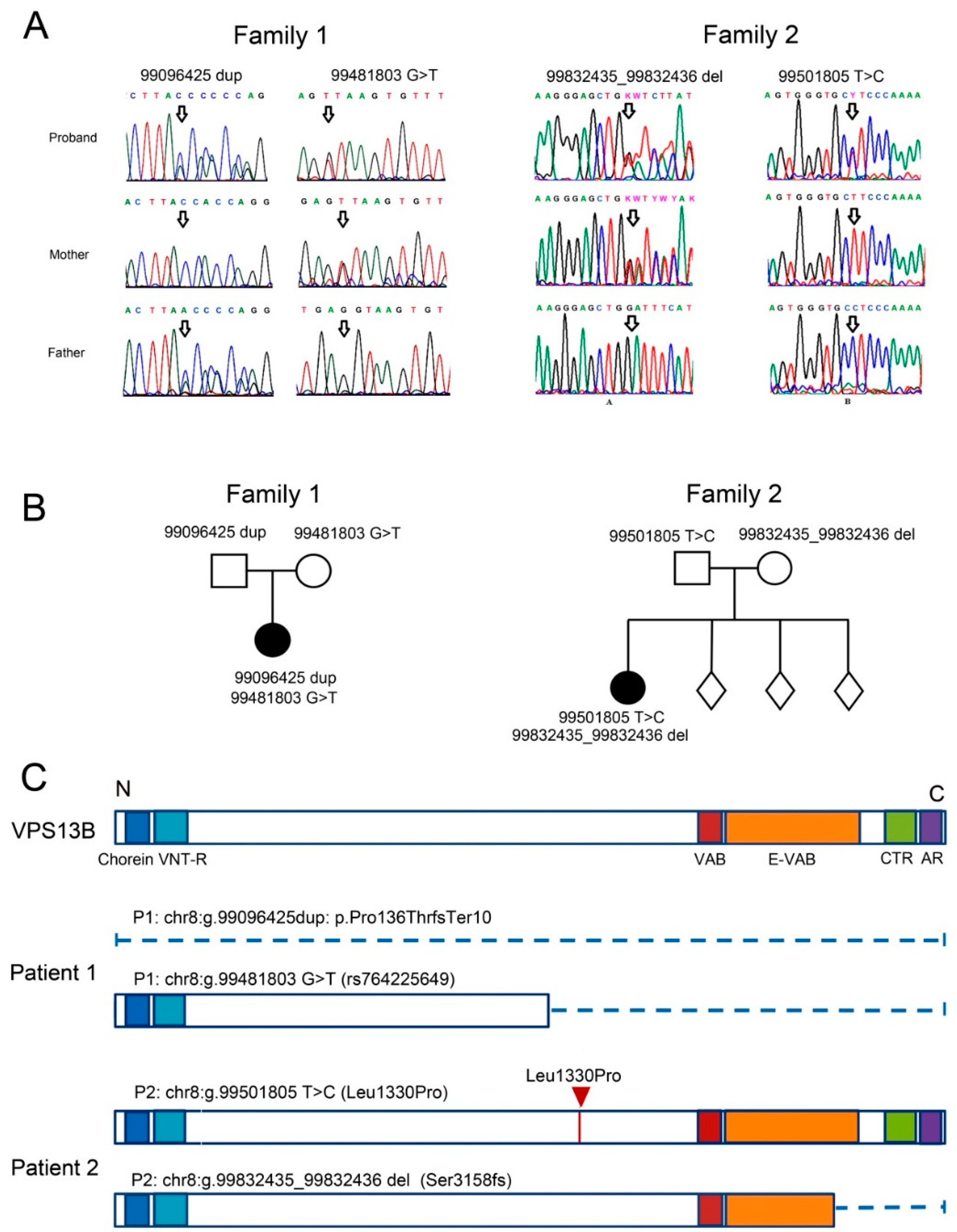
3.1. Case Report of Two Patients with CS and Mutations in the VPS13B Gene
3.1.1. Patient 1
3.1.2. Patient 2
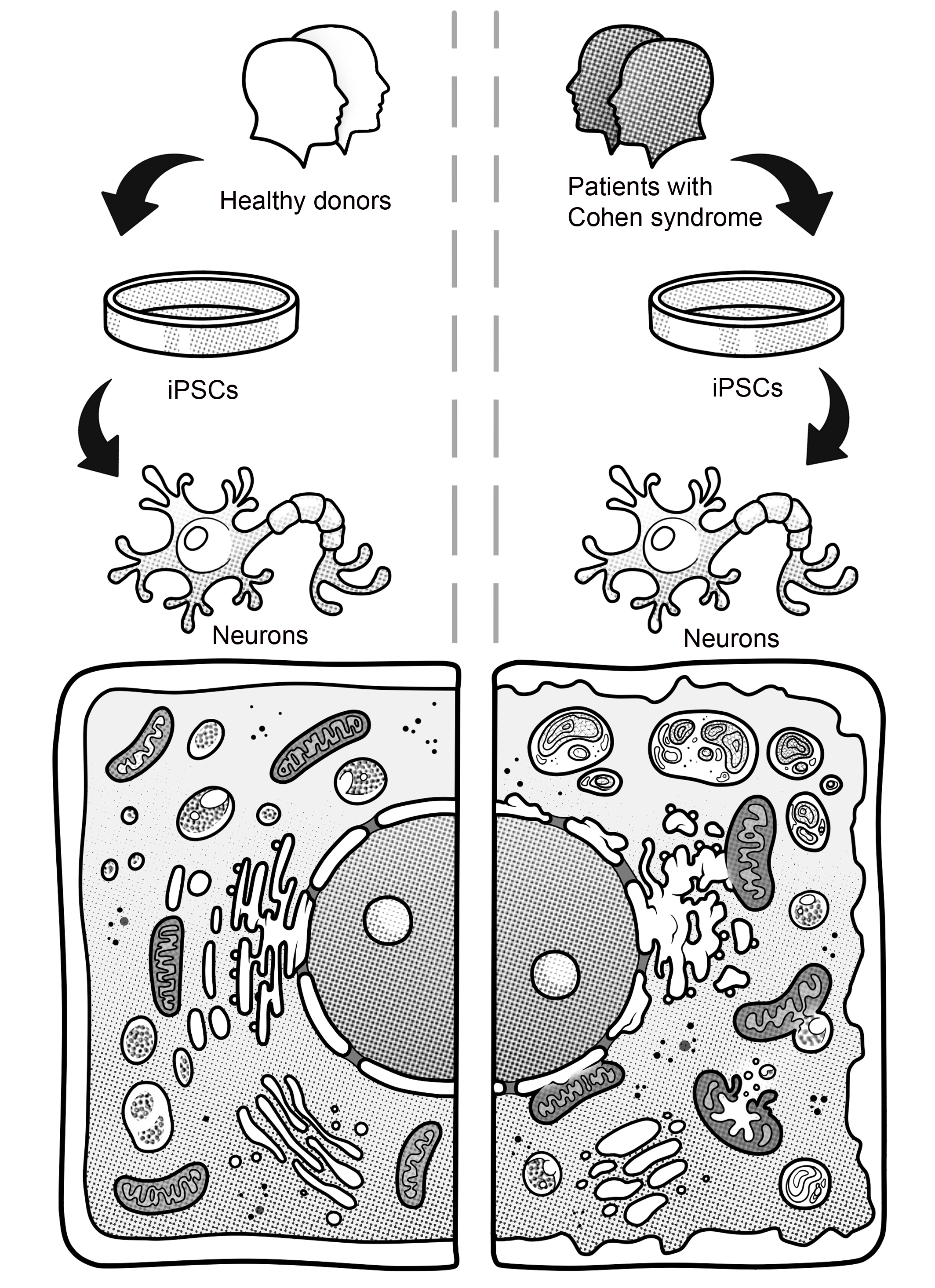
3.2. Generation of iPSCs for the CS Patients and Healthy Donors
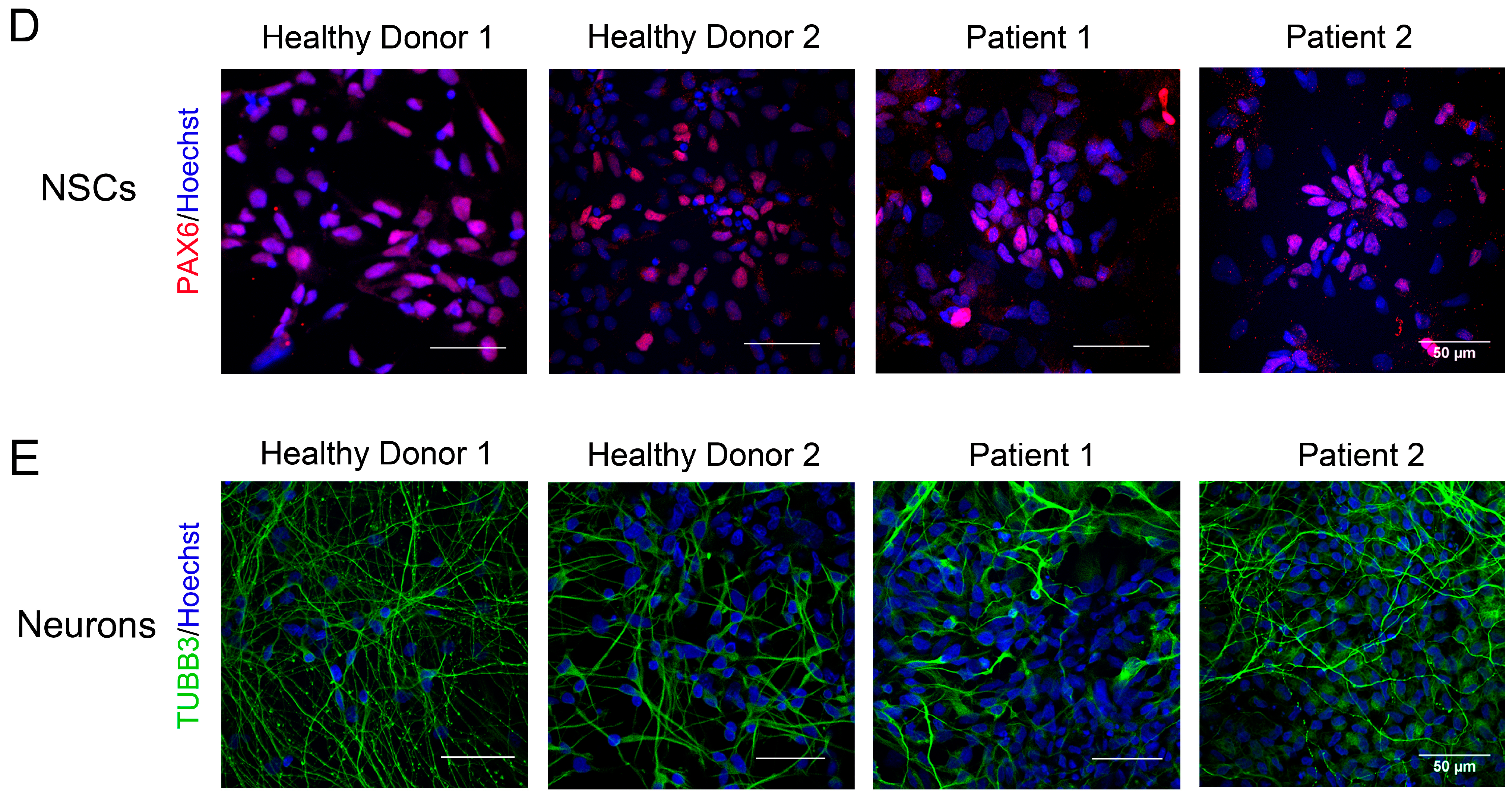
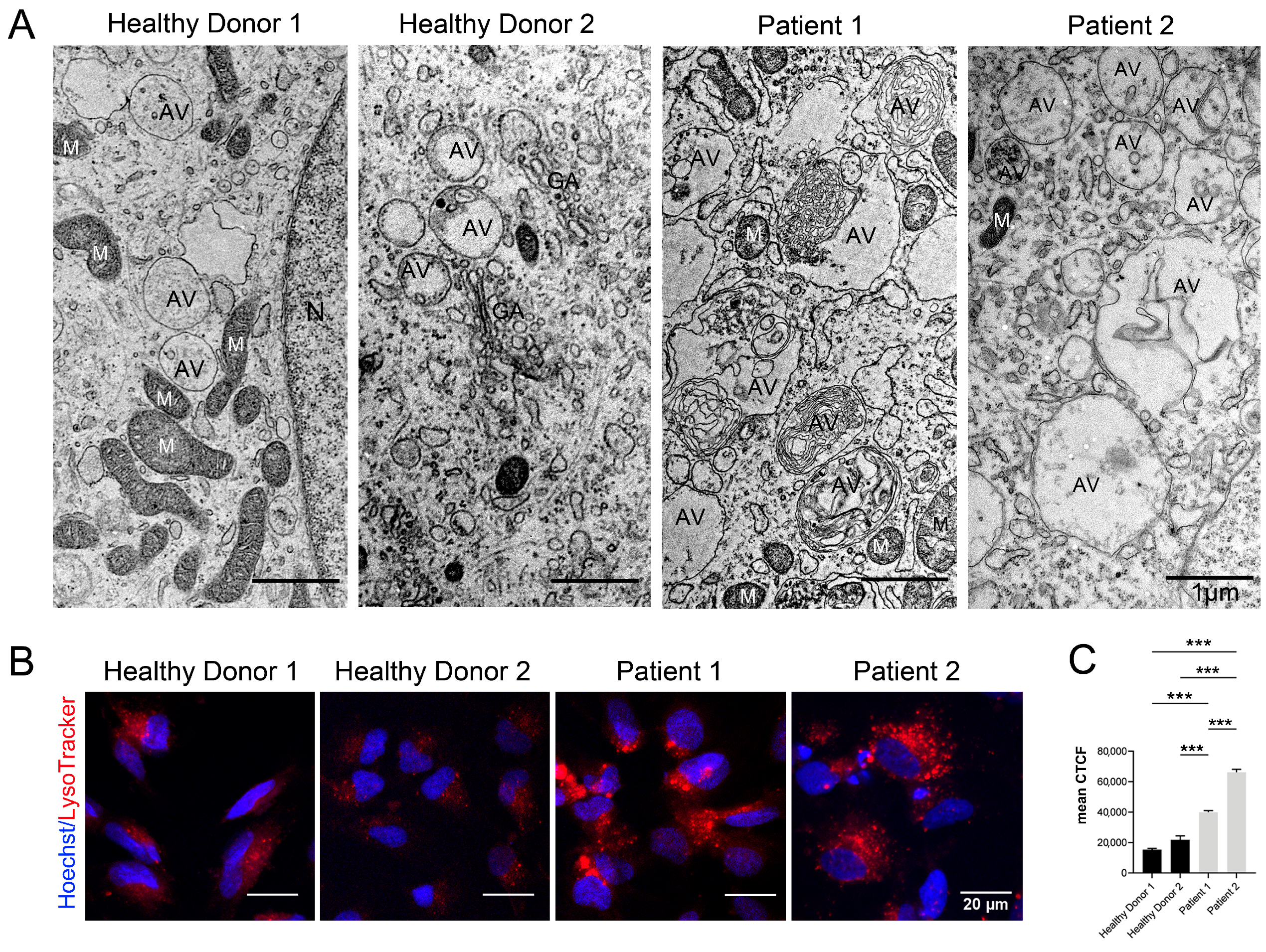
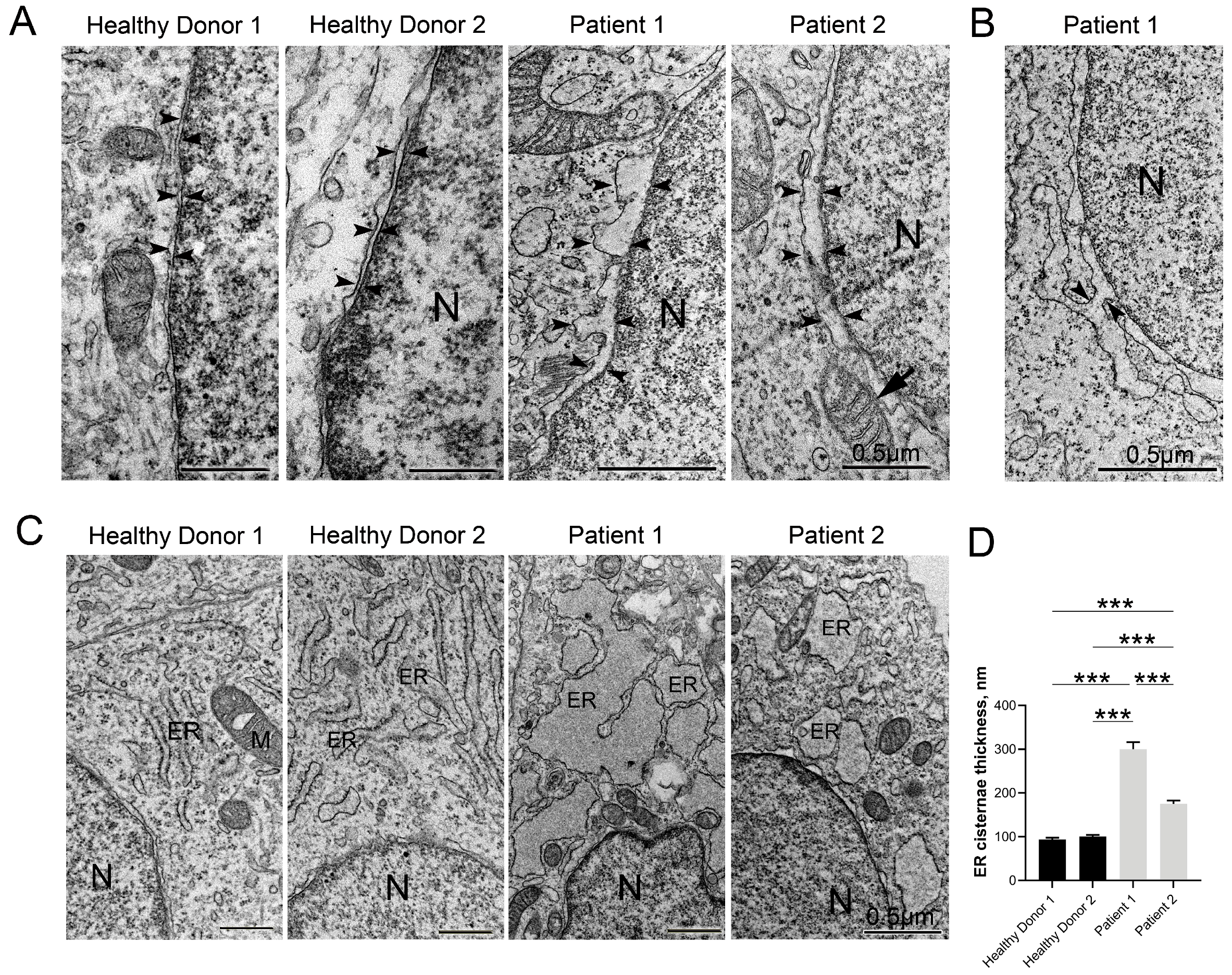
3.3. Immunocytological and Ultrastructural Analysis of CS Neuronal Cells
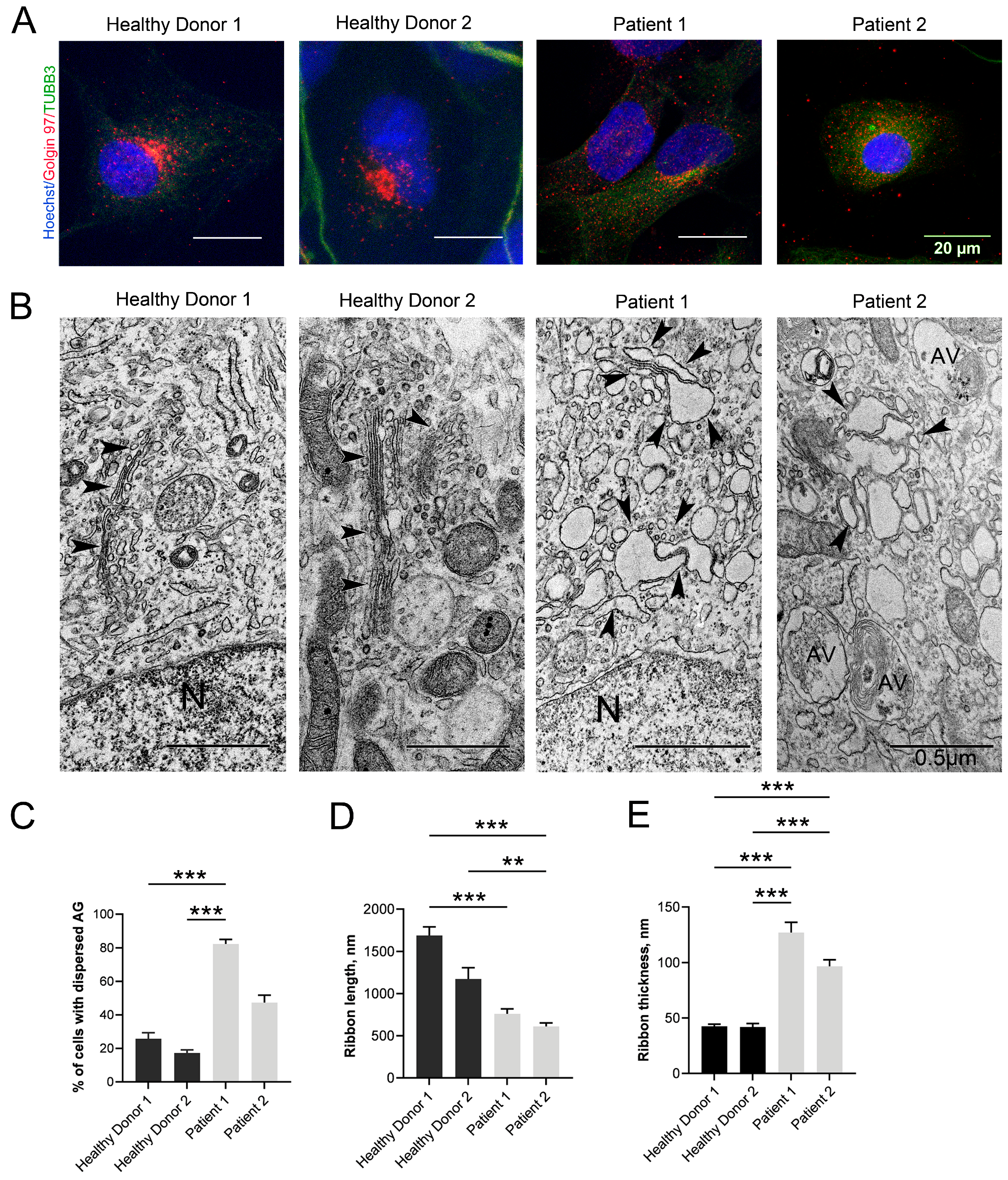
3.4. Changes in the Structure of the GA
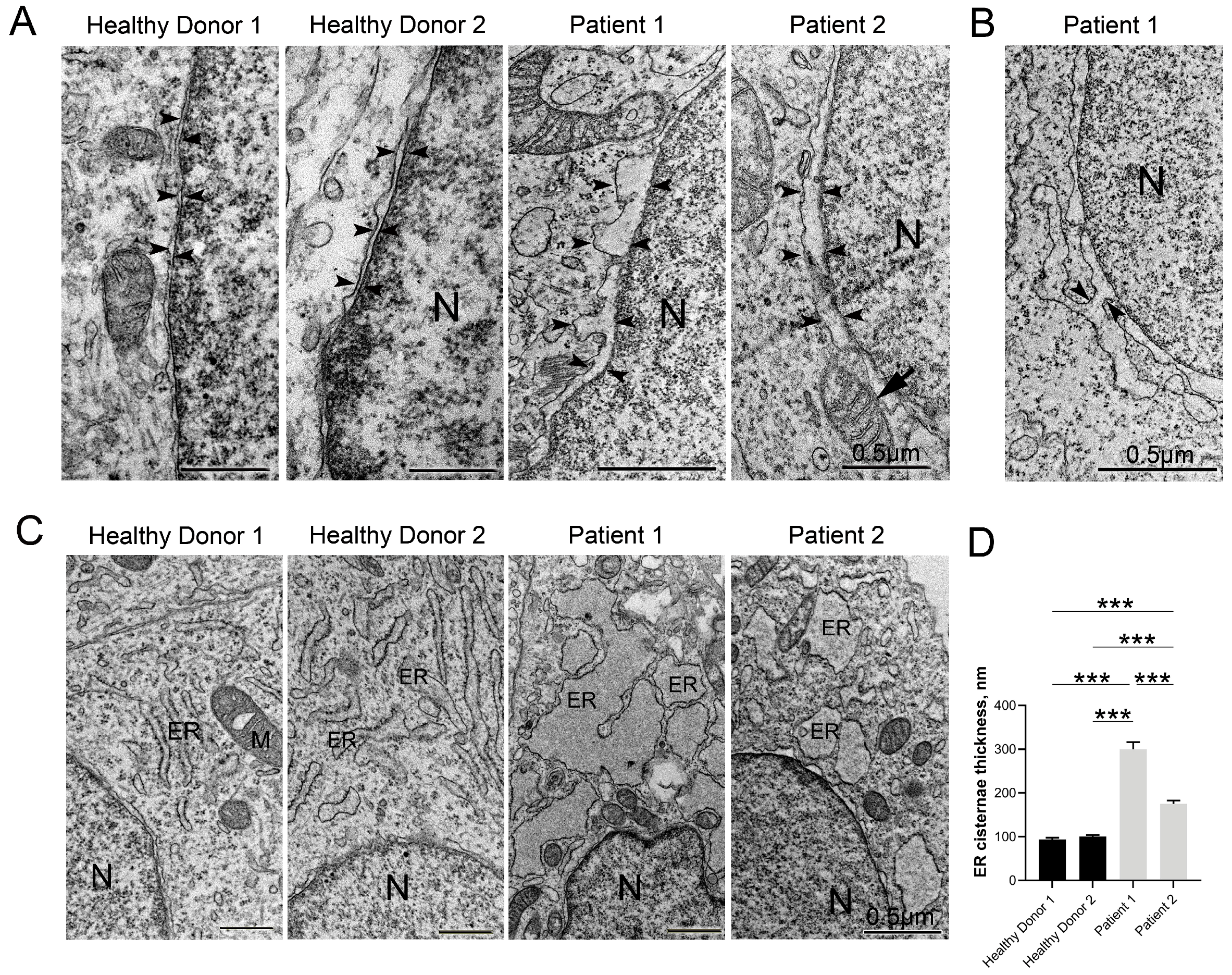
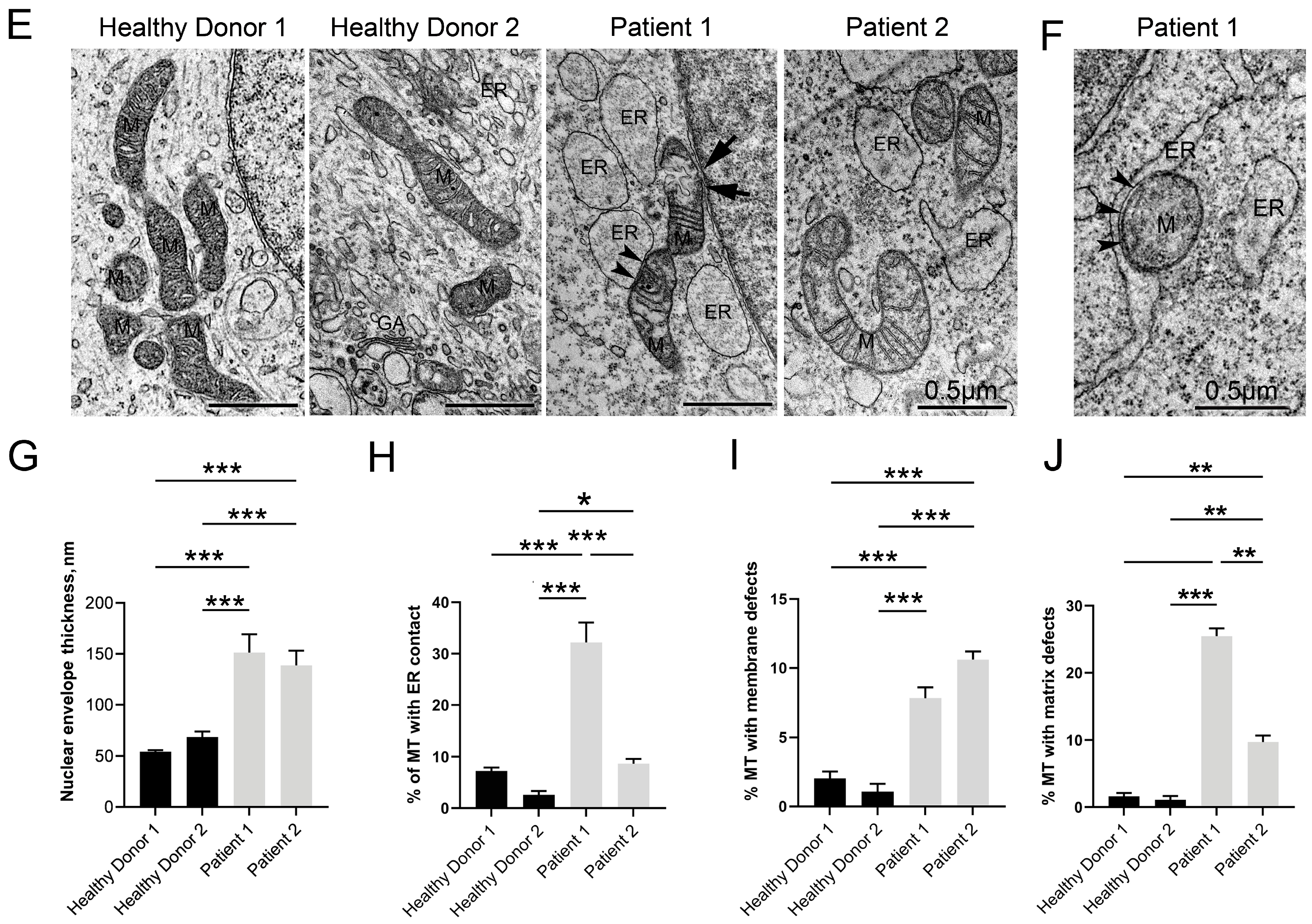
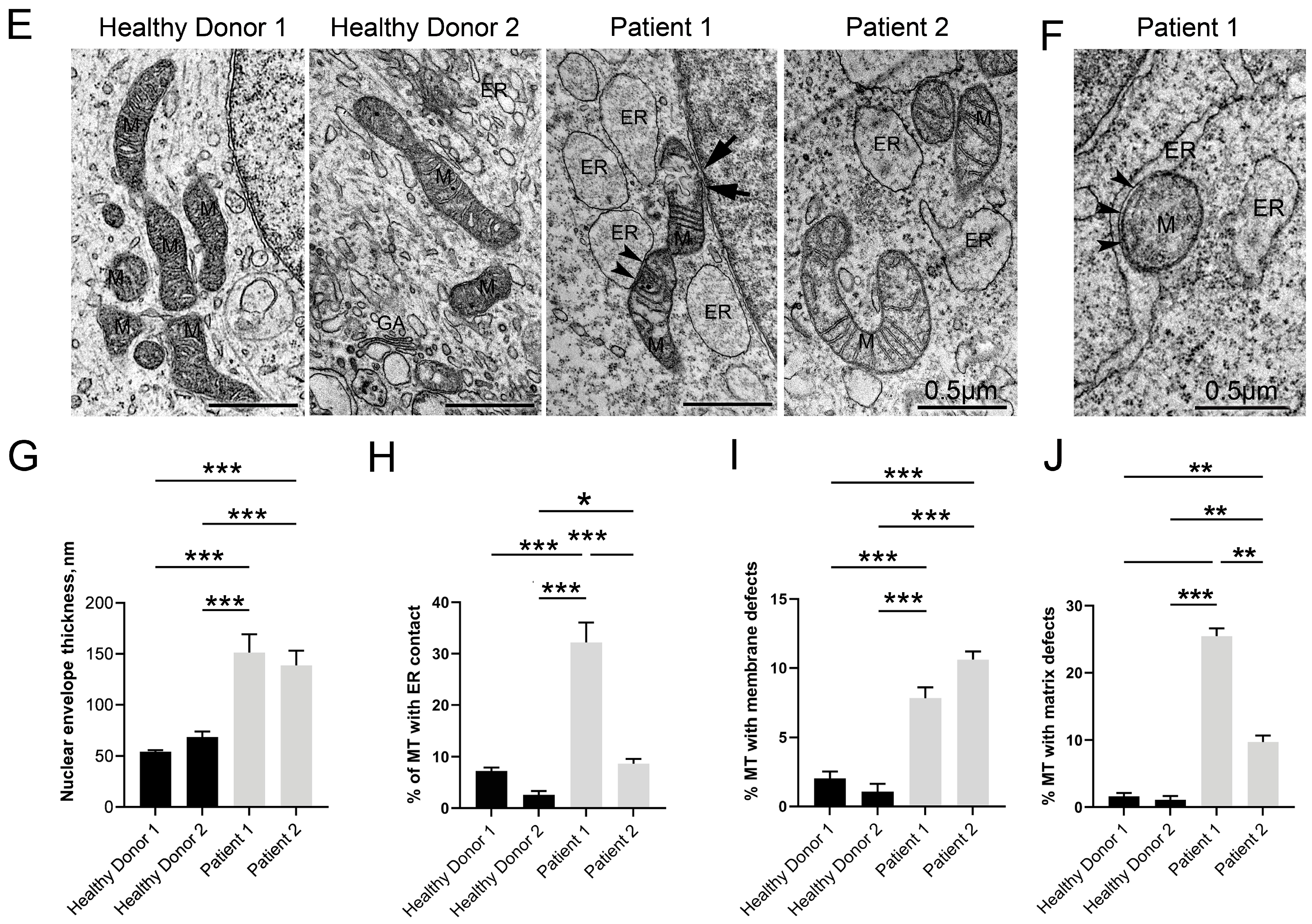
3.5. Disruptive Changes in the Structure of Other Cell Organelles
3.6. Autophagy Hallmarks in CS Patient-Derived Neuronal Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michael Cohen, M.; Hall, B.D.; Smith, D.W.; Benjamin Graham, C.; Lampert, K.J. A New Syndrome with Hypotonia, Obesity, Mental Deficiency, and Facial, Oral, Ocular, and Limb Anomalies. J. Pediatr. 1973, 83, 280–284. [Google Scholar] [CrossRef] [PubMed]
- El Chehadeh-Djebbar, S.; Blair, E.; Holder-Espinasse, M.; Moncla, A.; Frances, A.-M.; Rio, M.; Debray, F.-G.; Rump, P.; Masurel-Paulet, A.; Gigot, N.; et al. Changing Facial Phenotype in Cohen Syndrome: Towards Clues for an Earlier Diagnosis. Eur. J. Hum. Genet. 2013, 21, 736–742. [Google Scholar] [CrossRef]
- Güneş, N.; Alkaya, D.U.; Demirbilek, V.; Yalçınkaya, C.; Tüysüz, B. Early Diagnostic Signs and the Natural History of Typical Findings in Cohen Syndrome. J. Pediatr. 2023, 252, 93–100. [Google Scholar] [CrossRef]
- Rim, P.H.H.; Figueirêdo, E.S.d.; Hirata, F.E.; Steiner, C.E.; Marques-de-Faria, A.P. Ocular Findings in Brazilian Identical Twins with Cohen Syndrome: Case Report. Arq. Bras. Oftalmol. 2009, 72, 815–818. [Google Scholar] [CrossRef]
- Yang, C.; Hou, M.; Li, Y.; Sun, D.; Guo, Y.; Liu, P.; Liu, Y.; Song, J.; Zhang, N.; Wei, W.; et al. Gene Analysis: A Rare Gene Disease of Intellectual Deficiency-Cohen Syndrome. Int. J. Dev. Neurosci. 2018, 68, 83–88. [Google Scholar] [CrossRef]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Coban Akdemir, Z.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes That Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef]
- Rafiq, M.A.; Leblond, C.S.; Saqib, M.A.N.; Vincent, A.K.; Ambalavanan, A.; Khan, F.S.; Ayaz, M.; Shaheen, N.; Spiegelman, D.; Ali, G.; et al. Novel VPS13B Mutations in Three Large Pakistani Cohen Syndrome Families Suggests a Baloch Variant with Autistic-Like Features. BMC Med. Genet. 2015, 16, 41. [Google Scholar] [CrossRef]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef]
- Alipour, N.; Salehpour, S.; Tonekaboni, S.H.; Rostami, M.; Bahari, S.; Yassaee, V.; Miryounesi, M.; Ghafouri-Fard, S. Mutations in the VPS13B Gene in Iranian Patients with Different Phenotypes of Cohen Syndrome. J. Mol. Neurosci. MN 2020, 70, 21–25. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Hwang, S.-K.; Lee, S.-K.; Yang, J.; Kwak, J.-H.; Seo, H.; Ahn, H.; Lee, Y.-S.; Kim, J.; Lim, C.-S.; et al. Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes. J. Clin. Med. 2020, 9, 1886. [Google Scholar] [CrossRef]
- Nasser, F.; Kurtenbach, A.; Biskup, S.; Weidensee, S.; Kohl, S.; Zrenner, E. Ophthalmic Features of Retinitis Pigmentosa in Cohen Syndrome Caused by Pathogenic Variants in the VPS13B Gene. Acta Ophthalmol. 2020, 98, e316–e321. [Google Scholar] [CrossRef] [PubMed]
- North, K.N.; Fulton, A.B.; Whiteman, D.A.H. Identical Twins with Cohen Syndrome. Am. J. Med. Genet. 1995, 58, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Rejeb, I.; Jilani, H.; Elaribi, Y.; Hizem, S.; Hila, L.; Zillahrdt, J.L.; Chelly, J.; Benjemaa, L. First Case Report of Cohen Syndrome in the Tunisian Population Caused by VPS13B Mutations. BMC Med. Genet. 2017, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Hoyer, J.; Guth, S.; Zweier, C.; Kraus, C.; Becker, C.; Zenker, M.; Hüffmeier, U.; Thiel, C.; Rüschendorf, F.; et al. Diagnostic Yield of Various Genetic Approaches in Patients with Unexplained Developmental Delay or Mental Retardation. Am. J. Med. Genet. A 2006, 140A, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Kolehmainen, J.; Black, G.C.M.; Saarinen, A.; Chandler, K.; Clayton-Smith, J.; Träskelin, A.-L.; Perveen, R.; Kivitie-Kallio, S.; Norio, R.; Warburg, M.; et al. Cohen Syndrome Is Caused by Mutations in a Novel Gene, COH1, Encoding a Transmembrane Protein with a Presumed Role in Vesicle-Mediated Sorting and Intracellular Protein Transport. Am. J. Hum. Genet. 2003, 72, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Seifert, W.; Kühnisch, J.; Maritzen, T.; Horn, D.; Haucke, V.; Hennies, H.C. Cohen Syndrome-Associated Protein, COH1, Is a Novel, Giant Golgi Matrix Protein Required for Golgi Integrity*. J. Biol. Chem. 2011, 286, 37665–37675. [Google Scholar] [CrossRef] [PubMed]
- Seifert, W.; Holder-Espinasse, M.; Spranger, S.; Hoeltzenbein, M.; Rossier, E.; Dollfus, H.; Lacombe, D.; Verloes, A.; Chrzanowska, K.H.; Maegawa, G.H.B.; et al. Mutational Spectrum of COH1 and Clinical Heterogeneity in Cohen Syndrome. J. Med. Genet. 2006, 43, e22. [Google Scholar] [CrossRef]
- Boschann, F.; Fischer-Zirnsak, B.; Wienker, T.F.; Holtgrewe, M.; Seelow, D.; Eichhorn, B.; Döhnert, S.; Fahsold, R.; Horn, D.; Graul-Neumann, L.M. An Intronic Splice Site Alteration in Combination with a Large Deletion Affecting VPS13B (COH1) Causes Cohen Syndrome. Eur. J. Med. Genet. 2020, 63, 103973. [Google Scholar] [CrossRef]
- Zhao, S.; Luo, Z.; Xiao, Z.; Li, L.; Zhao, R.; Yang, Y.; Zhong, Y. Case Report: Two Novel VPS13B Mutations in a Chinese Family with Cohen Syndrome and Hyperlinear Palms. BMC Med. Genet. 2019, 20, 187. [Google Scholar] [CrossRef]
- Zorn, M.; Kühnisch, J.; Bachmann, S.; Seifert, W. Disease Relevance of Rare VPS13B Missense Variants for Neurodevelopmental Cohen Syndrome. Sci. Rep. 2022, 12, 9686. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C Are Lipid Transport Proteins Differentially Localized at ER Contact Sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Ryan, K.M. ATG2 and VPS13 Proteins: Molecular Highways Transporting Lipids to Drive Membrane Expansion and Organelle Communication. FEBS J. 2022, 289, 7113–7127. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Braceras, S.; Calvo, R.; Escalante, R. TipC and the Chorea-Acanthocytosis Protein VPS13A Regulate Autophagy in Dictyostelium and Human HeLa Cells. Autophagy 2015, 11, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Kotani, T.; Kawaoka, T.; Hirata, E.; Suzuki, K.; Nakatogawa, H.; Ohsumi, Y.; Noda, N.N. Atg2 Mediates Direct Lipid Transfer between Membranes for Autophagosome Formation. Nat. Struct. Mol. Biol. 2019, 26, 281–288. [Google Scholar] [CrossRef]
- Rampoldi, L.; Dobson-Stone, C.; Rubio, J.P.; Danek, A.; Chalmers, R.M.; Wood, N.W.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; et al. A Conserved Sorting-Associated Protein Is Mutant in Chorea-Acanthocytosis. Nat. Genet. 2001, 28, 119–120. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef]
- Gauthier, J.; Meijer, I.A.; Lessel, D.; Mencacci, N.E.; Krainc, D.; Hempel, M.; Tsiakas, K.; Prokisch, H.; Rossignol, E.; Helm, M.H.; et al. Recessive Mutations in VPS13D Cause Childhood Onset Movement Disorders. Ann. Neurol. 2018, 83, 1089–1095. [Google Scholar] [CrossRef]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.-J.; Trinh, J.; Brüggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D Lead to a New Recessive Ataxia with Spasticity and Mitochondrial Defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef]
- Dziurdzik, S.K.; Conibear, E. The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites. Int. J. Mol. Sci. 2021, 22, 2905. [Google Scholar] [CrossRef]
- Koike, S.; Jahn, R. SNAREs Define Targeting Specificity of Trafficking Vesicles by Combinatorial Interaction with Tethering Factors. Nat. Commun. 2019, 10, 1608. [Google Scholar] [CrossRef]
- Du, Y.; Xiong, J.; Ji, W.-K. Trans-Golgi Network-Lipid Droplet Contacts Maintain the TGN Integrity and Function via Lipid Transfer Activities of VPS13B. bioRxiv 2020. [Google Scholar]
- Li, P.; Lees, J.A.; Lusk, C.P.; Reinisch, K.M. Cryo-EM Reconstruction of a VPS13 Fragment Reveals a Long Groove to Channel Lipids between Membranes. J. Cell Biol. 2020, 219, e202001161. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, R.; Bordessoules, M.; Guilleman, M.; Carmignac, V.; Lhussiez, V.; Courot, H.; Bataille, A.; Chlémaire, A.; Bruno, C.; Fauque, P.; et al. Vps13b Is Required for Acrosome Biogenesis through Functions in Golgi Dynamic and Membrane Trafficking. Cell. Mol. Life Sci. 2020, 77, 511–529. [Google Scholar] [CrossRef]
- Seifert, W.; Kühnisch, J.; Maritzen, T.; Lommatzsch, S.; Hennies, H.C.; Bachmann, S.; Horn, D.; Haucke, V. Cohen Syndrome-Associated Protein COH1 Physically and Functionally Interacts with the Small GTPase RAB6 at the Golgi Complex and Directs Neurite Outgrowth*. J. Biol. Chem. 2015, 290, 3349–3358. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-K.; Lee, S.-K.; Choi, S.; Huh, Y.H.; Kwak, J.-H.; Lee, Y.-S.; Jang, D.-J.; Lee, J.-H.; Lee, K.; Kaang, B.-K.; et al. Autophagy Pathway Upregulation in a Human iPSC-Derived Neuronal Model of Cohen Syndrome with VPS13B Missense Mutations. Mol. Brain 2020, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.F.; Halloran, M.C. Centrosome Movements in Vivo Correlate with Specific Neurite Formation Downstream of LIM Homeodomain Transcription Factor Activity. Development 2012, 139, 3590–3599. [Google Scholar] [CrossRef]
- Cáceres, A.; Paglini, G.; Quiroga, S.; Ferreira, A. Role of the Golgi Apparatus During Axon Formation. In Intracellular Mechanisms for Neuritogenesis; de Curtis, I., Ed.; Springer US: Boston, MA, USA, 2007; pp. 136–154. ISBN 978-0-387-68561-8. [Google Scholar]
- Villarroel-Campos, D.; Gastaldi, L.; Conde, C.; Caceres, A.; Gonzalez-Billault, C. Rab-Mediated Trafficking Role in Neurite Formation. J. Neurochem. 2014, 129, 240–248. [Google Scholar] [CrossRef]
- Ori-McKenney, K.M.; Jan, L.Y.; Jan, Y.-N. Golgi Outposts Shape Dendrite Morphology by Functioning as Sites of Acentrosomal Microtubule Nucleation in Neurons. Neuron 2012, 76, 921–930. [Google Scholar] [CrossRef]
- Bogomiakova, M.E.; Sekretova, E.K.; Anufrieva, K.S.; Khabarova, P.O.; Kazakova, A.N.; Bobrovsky, P.A.; Grigoryeva, T.V.; Eremeev, A.V.; Lebedeva, O.S.; Bogomazova, A.N.; et al. iPSC-Derived Cells Lack Immune Tolerance to Autologous NK-Cells Due to Imbalance in Ligands for Activating and Inhibitory NK-Cell Receptors. Stem Cell Res. Ther. 2023, 14, 77. [Google Scholar] [CrossRef]
- Riedhammer, C.; Halbritter, D.; Weissert, R. Peripheral Blood Mononuclear Cells: Isolation, Freezing, Thawing, and Culture. In Multiple Sclerosis: Methods and Protocols; Weissert, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 53–61. ISBN 978-1-4939-2630-5. [Google Scholar]
- Grigor’eva, E.V.; Drozdova, E.S.; Sorogina, D.A.; Malakhova, A.A.; Pavlova, S.V.; Vyatkin, Y.V.; Khabarova, E.A.; Rzaev, J.A.; Medvedev, S.P.; Zakian, S.M. Generation of Induced Pluripotent Stem Cell Line, ICGi034-A, by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Parkinson’s Disease Associated with GBA Mutation. Stem Cell Res. 2022, 59, 102651. [Google Scholar] [CrossRef]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of Human ES and iPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Pisal, R.V.; Hrebíková, H.; Chvátalová, J.; Kunke, D.; Filip, S.; Mokrý, J. Detection of Mycoplasma Contamination Directly from Culture Supernatant Using Polymerase Chain Reaction. Folia Biol. 2016, 62, 203–206. [Google Scholar]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed Differentiation of Human Pluripotent Stem Cells to Cerebral Cortex Neurons and Neural Networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef]
- Morozova, K.N.; Suldina, L.A.; Malankhanova, T.B.; Grigor’eva, E.V.; Zakian, S.M.; Kiseleva, E.; Malakhova, A.A. Introducing an Expanded CAG Tract into the Huntingtin Gene Causes a Wide Spectrum of Ultrastructural Defects in Cultured Human Cells. PLoS ONE 2018, 13, e0204735. [Google Scholar] [CrossRef]
- Prokhorovich, M.A.; Lagar’kova, M.A.; Shilov, A.G.; Karamysheva, T.V.; Kiselyov, S.L.; Rubtsov, N.B. Cultures of hESM Human Embryonic Stem Cells: Chromosomal Aberrations and Karyotype Stability. Bull. Exp. Biol. Med. 2007, 144, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Measuring Cell Fluorescence Using ImageJ—The Open Lab Book v1.0. Available online: https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html (accessed on 4 October 2023).
- Leonzino, M.; Reinisch, K.M.; De Camilli, P. Insights into VPS13 Properties and Function Reveal a New Mechanism of Eukaryotic Lipid Transport. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2021, 1866, 159003. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Mailliot, J.; Rigger, L.; Neuner, S.; Shin, B.-S.; Yusupova, G.; Dever, T.E.; Micura, R.; Yusupov, M. Molecular Insights into Protein Synthesis with Proline Residues. EMBO Rep. 2016, 17, 1776–1784. [Google Scholar] [CrossRef]
- Gridina, M.M.; Matveeva, N.M.; Fishman, V.S.; Menzorov, A.G.; Kizilova, H.A.; Beregovoy, N.A.; Kovrigin, I.I.; Pristyazhnyuk, I.E.; Oscorbin, I.P.; Filipenko, M.L.; et al. Allele-Specific Biased Expression of the CNTN6 Gene in iPS Cell-Derived Neurons from a Patient with Intellectual Disability and 3p26.3 Microduplication Involving the CNTN6 Gene. Mol. Neurobiol. 2018, 55, 6533–6546. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-Associated Membranes as Hubs for Neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Flock, D.L.; Martin, L.J.; Northington, F.J. Endoplasmic Reticulum Pathology and Stress Response in Neurons Precede Programmed Necrosis after Neonatal Hypoxia-Ischemia. Int. J. Dev. Neurosci. 2016, 48, 58–70. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode)Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Degechisa, S.T.; Dabi, Y.T.; Gizaw, S.T. The Mitochondrial Associated Endoplasmic Reticulum Membranes: A Platform for the Pathogenesis of Inflammation-Mediated Metabolic Diseases. Immun. Inflamm. Dis. 2022, 10, e647. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with My MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, R.U.; Oh, J.; Choi, J.E.; Kim, H.; Lee, K.; Hwang, S.-K.; Lee, J.-H.; Lee, J.-A.; Kaang, B.-K.; et al. Spatial Learning and Motor Deficits in Vacuolar Protein Sorting-Associated Protein 13b (Vps13b) Mutant Mouse. Exp. Neurobiol. 2019, 28, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Shearman, J.R.; Wilton, A.N. A Canine Model of Cohen Syndrome: Trapped Neutrophil Syndrome. BMC Genom. 2011, 12, 258. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Duplomb, L.; Duvet, S.; Picot, D.; Jego, G.; El Chehadeh-Djebbar, S.; Marle, N.; Gigot, N.; Aral, B.; Carmignac, V.; Thevenon, J.; et al. Cohen Syndrome Is Associated with Major Glycosylation Defects. Hum. Mol. Genet. 2014, 23, 2391–2399. [Google Scholar] [CrossRef]
- Sütterlin, C.; Hsu, P.; Mallabiabarrena, A.; Malhotra, V. Fragmentation and Dispersal of the Pericentriolar Golgi Complex Is Required for Entry into Mitosis in Mammalian Cells. Cell 2002, 109, 359–369. [Google Scholar] [CrossRef]
- Wei, J.-H.; Seemann, J. Unraveling the Golgi Ribbon. Traffic 2010, 11, 1391–1400. [Google Scholar] [CrossRef]
- Wei, J.-H.; Seemann, J. Golgi Ribbon Disassembly during Mitosis, Differentiation and Disease Progression. Curr. Opin. Cell Biol. 2017, 47, 43–51. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Li, T.; Jiang, Z.; Zeng, L.; Hu, Z. The Role of the Golgi Apparatus in Disease (Review). Int. J. Mol. Med. 2021, 47, 38. [Google Scholar] [CrossRef] [PubMed]
- Haase, G.; Rabouille, C. Golgi Fragmentation in ALS Motor Neurons. New Mechanisms Targeting Microtubules, Tethers, and Transport Vesicles. Front. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Haukedal, H.; Corsi, G.I.; Gadekar, V.P.; Doncheva, N.T.; Kedia, S.; de Haan, N.; Chandrasekaran, A.; Jensen, P.; Schiønning, P.; Vallin, S.; et al. Golgi Fragmentation—One of the Earliest Organelle Phenotypes in Alzheimer’s Disease Neurons. Front. Neurosci. 2023, 17, 1120086. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Bekier, M.; Wang, Y. Golgi Fragmentation in Alzheimer’s Disease. Front. Neurosci. 2015, 9, 340. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.Á.; Tomás, M.; Martínez-Martínez, N.; Martínez-Alonso, E. Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef]
- Tomás, M.; Martínez-Alonso, E.; Martínez-Martínez, N.; Cara-Esteban, M.; Martínez-Menárguez, J.A. Fragmentation of the Golgi Complex of Dopaminergic Neurons in Human Substantia Nigra: New Cytopathological Findings in Parkinson’s Disease. Histol. Histopathol. 2021, 36, 47–60. [Google Scholar] [CrossRef]
- Gosavi, P.; Houghton, F.J.; McMillan, P.J.; Hanssen, E.; Gleeson, P.A. The Golgi Ribbon in Mammalian Cells Negatively Regulates Autophagy by Modulating mTOR Activity. J. Cell Sci. 2018, 131, jcs211987. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Stone, M.C.; Rolls, M.M. Microtubules Are Organized Independently of the Centrosome in Drosophilaneurons. Neural Dev. 2011, 6, 38. [Google Scholar] [CrossRef]
- Rios, R.M. The Centrosome–Golgi Apparatus Nexus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130462. [Google Scholar] [CrossRef]
- Stiess, M.; Maghelli, N.; Kapitein, L.C.; Gomis-Rüth, S.; Wilsch-Bräuninger, M.; Hoogenraad, C.C.; Tolić-Nørrelykke, I.M.; Bradke, F. Axon Extension Occurs Independently of Centrosomal Microtubule Nucleation. Science 2010, 327, 704–707. [Google Scholar] [CrossRef]
- Guadagno, N.A.; Progida, C. Rab GTPases: Switching to Human Diseases. Cells 2019, 8, 909. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, P.-S.; Yang, L. Golgi-Associated Rab GTPasesimplicated in Autophagy. Cell Biosci. 2021, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Ayala, C.I.; Kim, J.; Neufeld, T.P. Rab6 Promotes Insulin Receptor and Cathepsin Trafficking to Regulate Autophagy Induction and Activity in Drosophila. J. Cell Sci. 2018, 131, jcs216127. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.R.; Gahl, W.A. Disorders of Metal Metabolism. Transl. Sci. Rare Dis. 2017, 2, 101–139. [Google Scholar] [CrossRef]
- Bennett, M.J.; Rakheja, D. The Neuronal Ceroid-Lipofuscinoses. Dev. Disabil. Res. Rev. 2013, 17, 254–259. [Google Scholar] [CrossRef]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal Dominant Neuronal Ceroid Lipofuscinosis: Clinical Features and Molecular Basis. Clin. Genet. 2021, 99, 111–118. [Google Scholar] [CrossRef]
- Vu, M.; Li, R.; Baskfield, A.; Lu, B.; Farkhondeh, A.; Gorshkov, K.; Motabar, O.; Beers, J.; Chen, G.; Zou, J.; et al. Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Tay-Sachs Disease. Orphanet J. Rare Dis. 2018, 13, 152. [Google Scholar] [CrossRef]
- Kirkbride, K.C.; Hong, N.H.; French, C.L.; Clark, E.S.; Jerome, W.G.; Weaver, A.M. Regulation of Late Endosomal/Lysosomal Maturation and Trafficking by Cortactin Affects Golgi Morphology. Cytoskeleton 2012, 69, 625–643. [Google Scholar] [CrossRef]
- Roy, E.; Bruyère, J.; Flamant, P.; Bigou, S.; Ausseil, J.; Vitry, S.; Heard, J.M. GM130 Gain-of-Function Induces Cell Pathology in a Model of Lysosomal Storage Disease. Hum. Mol. Genet. 2012, 21, 1481–1495. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Brenman, J.E. Lysosomal Storage Diseases-Regulating Neurodegeneration. J. Exp. Neurosci. 2015, 9, 81–91. [Google Scholar] [CrossRef]
- Osellame, L.D.; Duchen, M.R. Quality Control Gone Wrong: Mitochondria, Lysosomal Storage Disorders and Neurodegeneration. Br. J. Pharmacol. 2014, 171, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; Sano, R.; d’Azzo, A. Mitochondria-Associated ER Membranes (MAMs) and Lysosomal Storage Diseases. Cell Death Dis. 2018, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-Ganglioside Accumulation at the Mitochondria-Associated ER Membranes Links ER Stress to Ca(2+)-Dependent Mitochondrial Apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic Reticulum Stress and Inflammation in the Central Nervous System. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Jang, W.; Puchkov, D.; Samsó, P.; Liang, Y.; Nadler-Holly, M.; Sigrist, S.J.; Kintscher, U.; Liu, F.; Mamchaoui, K.; Mouly, V.; et al. Endosomal Lipid Signaling Reshapes the Endoplasmic Reticulum to Control Mitochondrial Function. Science 2022, 378, eabq5209. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnaider, T.A.; Khabarova, A.A.; Morozova, K.N.; Yunusova, A.M.; Yakovleva, S.A.; Chvileva, A.S.; Wolf, E.R.; Kiseleva, E.V.; Grigor’eva, E.V.; Voinova, V.Y.; et al. Ultrastructural Abnormalities in Induced Pluripotent Stem Cell-Derived Neural Stem Cells and Neurons of Two Cohen Syndrome Patients. Cells 2023, 12, 2702. https://doi.org/10.3390/cells12232702
Shnaider TA, Khabarova AA, Morozova KN, Yunusova AM, Yakovleva SA, Chvileva AS, Wolf ER, Kiseleva EV, Grigor’eva EV, Voinova VY, et al. Ultrastructural Abnormalities in Induced Pluripotent Stem Cell-Derived Neural Stem Cells and Neurons of Two Cohen Syndrome Patients. Cells. 2023; 12(23):2702. https://doi.org/10.3390/cells12232702
Chicago/Turabian StyleShnaider, Tatiana A., Anna A. Khabarova, Ksenia N. Morozova, Anastasia M. Yunusova, Sophia A. Yakovleva, Anastasia S. Chvileva, Ekaterina R. Wolf, Elena V. Kiseleva, Elena V. Grigor’eva, Viktori Y. Voinova, and et al. 2023. "Ultrastructural Abnormalities in Induced Pluripotent Stem Cell-Derived Neural Stem Cells and Neurons of Two Cohen Syndrome Patients" Cells 12, no. 23: 2702. https://doi.org/10.3390/cells12232702
APA StyleShnaider, T. A., Khabarova, A. A., Morozova, K. N., Yunusova, A. M., Yakovleva, S. A., Chvileva, A. S., Wolf, E. R., Kiseleva, E. V., Grigor’eva, E. V., Voinova, V. Y., Lagarkova, M. A., Pomerantseva, E. A., Musatova, E. V., Smirnov, A. V., Smirnova, A. V., Stoklitskaya, D. S., Arefieva, T. I., Larina, D. A., Nikitina, T. V., & Pristyazhnyuk, I. E. (2023). Ultrastructural Abnormalities in Induced Pluripotent Stem Cell-Derived Neural Stem Cells and Neurons of Two Cohen Syndrome Patients. Cells, 12(23), 2702. https://doi.org/10.3390/cells12232702