
Stefin B Inhibits NLRP3 Inflammasome Activation via AMPK/mTOR Signalling
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Reagents
2.2. Mice
2.3. Preparation of Macrophages
2.4. Quantitative Real Time PCR
2.5. Cell Lysate Preparation and Western Blot Analysis
2.6. Cathepsin Activity Measurements
2.7. Cytokine Analysis
2.8. Nitric Oxide Measurements
2.9. Cytotoxicity, Lactate Dehydrogenase Release
2.10. Flow Cytometry
2.11. Confocal Microscopy
2.12. Analysis of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Using Seahorse XF Analyser
2.13. Statistical Analysis
3. Results
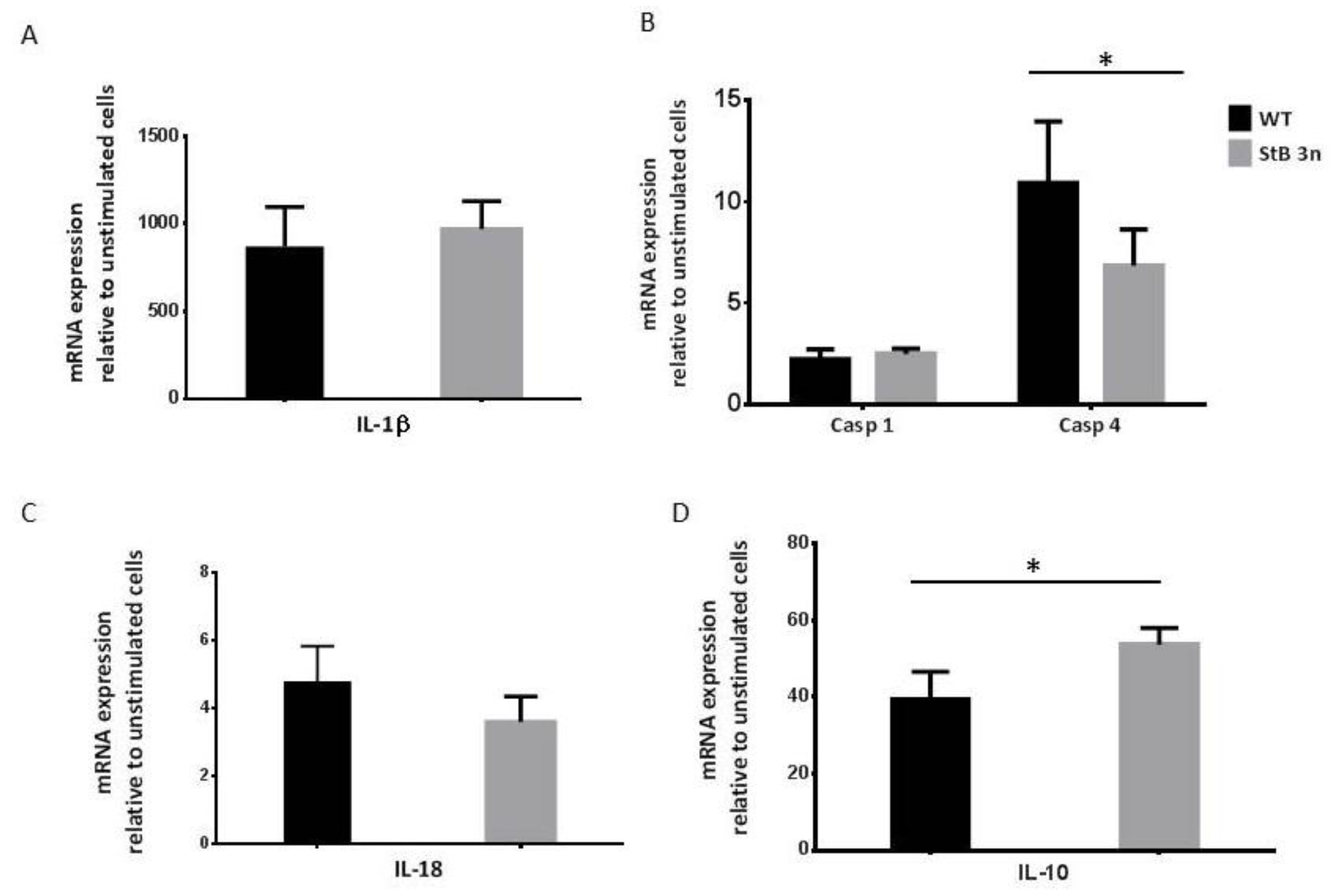
3.1. Additional Copy of Stefin B Downregulated Caspase-11 Expression Induced by LPS and NLRP3 Inflammasome Activation
3.2. Reduced Nitric Oxide (NO) Production in Stefin B 3n BMDMs Following IFN-γ and LPS Stimulation
3.3. Increased Stefin B Expression Prevents Accumulation of Dysfunctional Mitochondria
3.4. Increased Stefin B Expression Decreases OXPHOS Activity Following LPS Treatment
3.5. Stefin B Promotes the Induction of Mitophagy
3.6. Deregulated AMPK and mTORC1 Signalling in Stefin B 3n BMDMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sultan, M.; Piccini, I.; Balzereit, D.; Herwig, R.; Saran, N.G.; Lehrach, H.; Reeves, R.H.; Yaspo, M.-L. Gene expression variation in Down’s syndrome mice allows prioritization of candidate genes. Genome Biol. 2007, 8, R91. [Google Scholar] [CrossRef]
- Lehesjoki, A.E.; Kälviäinen, R. Progressive Myoclonic Epilepsy Type 1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pennacchio, L.A.; Lehesjoki, A.E.; Stone, N.E.; Willour, V.L.; Virtaneva, K.; Miao, J.; D’Amato, E.; Ramirez, L.; Faham, M.; Koskiniemi, M.; et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 1996, 271, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, T.; Kuronen, M.; Alakurtti, K.; Tegelberg, S.; Hakala, P.; Aalto, A.; Huopaniemi, L.; Aula, N.; Michellucci, R.; Eriksson, K.; et al. Cystatin B: Mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur. J. Hum. Genet. EJHG 2007, 15, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar] [PubMed]
- Pennacchio, L.A.; Bouley, D.M.; Higgins, K.M.; Scott, M.P.; Noebels, J.L.; Myers, R.M. Progressive ataxia, myoclonic epilepsy and cerebellar apoptosis in cystatin B-deficient mice. Nat. Genet. 1998, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Manninen, O.; Koskenkorva, P.; Lehtimäki, K.K.; Hyppönen, J.; Könönen, M.; Laitinen, T.; Kalimo, H.; Kopra, O.; Kälviäinen, R.; Gröhn, O.; et al. White matter degeneration with Unverricht-Lundborg progressive myoclonus epilepsy: A translational diffusion-tensor imaging study in patients and cystatin B-deficient mice. Radiology 2013, 269, 232–239. [Google Scholar] [CrossRef]
- Tegelberg, S.; Kopra, O.; Joensuu, T.; Cooper, J.D.; Lehesjoki, A.E. Early microglial activation precedes neuronal loss in the brain of the Cstb-/- mouse model of progressive myoclonus epilepsy, EPM1. J. Neuropathol. Exp. Neurol. 2012, 71, 40–53. [Google Scholar] [CrossRef]
- Ceru, S.; Konjar, S.; Maher, K.; Repnik, U.; Krizaj, I.; Bencina, M.; Renko, M.; Nepveu, A.; Zerovnik, E.; Turk, B.; et al. Stefin B interacts with histones and cathepsin L in the nucleus. J. Biol. Chem. 2010, 285, 10078–10086. [Google Scholar] [CrossRef] [PubMed]
- Daura, E.; Tegelberg, S.; Yoshihara, M.; Jackson, C.; Simonetti, F.; Aksentjeff, K.; Ezer, S.; Hakala, P.; Katayama, S.; Kere, J.; et al. Cystatin B-deficiency triggers ectopic histone H3 tail cleavage during neurogenesis. Neurobiol. Dis. 2021, 156, 105418. [Google Scholar] [CrossRef]
- Maher, K.; Završnik, J.; Jerič-Kokelj, B.; Vasiljeva, O.; Turk, B.; Kopitar-Jerala, N. Decreased IL-10 expression in stefin B-deficient macrophages is regulated by the MAP kinase and STAT-3 signaling pathways. FEBS Lett. 2014, 588, 720–726. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009, 29, 5910–5915. [Google Scholar] [CrossRef]
- Maher, K.; Jeric Kokelj, B.; Butinar, M.; Mikhaylov, G.; Mancek-Keber, M.; Stoka, V.; Vasiljeva, O.; Turk, B.; Grigoryev, S.A.; Kopitar-Jerala, N. A role for stefin B (cystatin B) in inflammation and endotoxemia. J. Biol. Chem. 2014, 289, 31736–31750. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. The Role of Stefin B in Neuro-inflammation. Front. Cell. Neurosci. 2015, 9, 458. [Google Scholar] [CrossRef]
- Okuneva, O.; Korber, I.; Li, Z.; Tian, L.; Joensuu, T.; Kopra, O.; Lehesjoki, A.E. Abnormal microglial activation in the Cstb(-/-) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia 2015, 63, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Okuneva, O.; Li, Z.; Korber, I.; Tegelberg, S.; Joensuu, T.; Tian, L.; Lehesjoki, A.E. Brain inflammation is accompanied by peripheral inflammation in Cstb (-/-) mice, a model for progressive myoclonus epilepsy. J. Neuroinflamm. 2016, 13, 298. [Google Scholar] [CrossRef] [PubMed]
- Brault, V.; Martin, B.; Costet, N.; Bizot, J.-C.; Hérault, Y. Characterization of PTZ-Induced Seizure Susceptibility in a Down Syndrome Mouse Model That Overexpresses CSTB. PLoS ONE 2011, 6, e27845. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Broz, P. Immunology: Caspase target drives pyroptosis. Nature 2015, 526, 642–643. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. 2019, 35, 453–475. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Biasizzo, M.; Trstenjak-Prebanda, M.; Dolinar, K.; Pirkmajer, S.; Završnik, J.; Turk, B.; Kopitar-Jerala, N. Cystatin C Deficiency Increases LPS-Induced Sepsis and NLRP3 Inflammasome Activation in Mice. Cells 2021, 10, 2071. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting Edge: Nitric Oxide Inhibits the NLRP3 Inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Trstenjak Prebanda, M.; Završnik, J.; Turk, B.; Kopitar Jerala, N. Upregulation of Mitochondrial Redox Sensitive Proteins in LPS-Treated Stefin B-Deficient Macrophages. Cells 2019, 8, 1476. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Barrett, A.J. Alpha 2-macroglobulin. Methods Enzym. 1981, 80, 737–754. [Google Scholar]
- Barrett, A.J.; Fritz, H.; Grubb, A.; Isemura, S.; Jarvinen, M.; Katunuma, N.; Machleidt, W.; Muller-Esterl, W.; Sasaki, M.; Turk, V. Nomenclature and classification of the proteins homologous with the cysteine-proteinase inhibitor chicken cystatin. Biochem. J. 1986, 236, 312. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. A J. Virtual Libr. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. The role of cystatins in cells of the immune system. FEBS Lett. 2006, 580, 6295–6301. [Google Scholar] [CrossRef]
- Magister, S.; Kos, J. Cystatins in immune system. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gren, S.T.; Janciauskiene, S.; Sandeep, S.; Jonigk, D.; Kvist, P.H.; Gerwien, J.G.; Håkansson, K.; Grip, O. The protease inhibitor cystatin C down-regulates the release of IL-β and TNF-α in lipopolysaccharide activated monocytes. J. Leukoc. Biol. 2016, 100, 811–822. [Google Scholar] [CrossRef]
- Rinne, R.; Saukko, P.; Jarvinen, M.; Lehesjoki, A.E. Reduced cystatin B activity correlates with enhanced cathepsin activity in progressive myoclonus epilepsy. Ann. Med. 2002, 34, 380–385. [Google Scholar] [CrossRef]
- Gorski, K.; Spoljaric, A.; Nyman, T.A.; Kaila, K.; Battersby, B.J.; Lehesjoki, A.E. Quantitative Changes in the Mitochondrial Proteome of Cerebellar Synaptosomes from Preclinical Cystatin B-Deficient Mice. Front. Mol. Neurosci. 2020, 13, 570640. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Kim, M.-J.; Bae, S.H.; Ryu, J.-C.; Kwon, Y.; Oh, J.-H.; Kwon, J.; Moon, J.-S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Pan, Y.; Shi, G.; Ren, J.; Fan, H.; Dou, H.; Hou, Y. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim. Biophys. Sin. 2018, 50, 888–896. [Google Scholar] [CrossRef]
- Tizon, B.; Sahoo, S.; Yu, H.; Gauthier, S.; Kumar, A.R.; Mohan, P.; Figliola, M.; Pawlik, M.; Grubb, A.; Uchiyama, Y.; et al. Induction of autophagy by cystatin C: A mechanism that protects murine primary cortical neurons and neuronal cell lines. PLoS ONE 2010, 5, 0009819. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sultana, N.; Siraj, N.; Ward, L.J.; Pawlik, M.; Levy, E.; Jovinge, S.; Bengtsson, E.; Yuan, X.M. Autophagy dysfunction and regulatory cystatin C in macrophage death of atherosclerosis. J. Cell. Mol. Med. 2016, 20, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Komine, O.; Endo, F.; Wakasugi, K.; Yamanaka, K. Intracerebroventricular administration of Cystatin C ameliorates disease in SOD1-linked amyotrophic lateral sclerosis mice. J. Neurochem. 2018, 145, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Orozco, J.M.; Adelmann, C.H.; Spinelli, J.B.; van der Helm, P.W.; Roberts, J.M.; Kunchok, T.; Sabatini, D.M. Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2021, 118, 2022120118. [Google Scholar] [CrossRef]
- Kaur, G.; Mohan, P.; Pawlik, M.; DeRosa, S.; Fajiculay, J.; Che, S.; Grubb, A.; Ginsberg, S.D.; Nixon, R.A.; Levy, E. Cystatin C rescues degenerating neurons in a cystatin B-knockout mouse model of progressive myoclonus epilepsy. Am. J. Pathol. 2010, 177, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N.; Curin-Serbec, V.; Jerala, R.; Krizaj, I.; Gubensek, F.; Turk, V. Monoclonal antibodies to human stefin B and determination of their epitopes. Biochim. Biophys. Acta 1993, 1164, 75–80. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trstenjak-Prebanda, M.; Biasizzo, M.; Dolinar, K.; Pirkmajer, S.; Turk, B.; Brault, V.; Herault, Y.; Kopitar-Jerala, N. Stefin B Inhibits NLRP3 Inflammasome Activation via AMPK/mTOR Signalling. Cells 2023, 12, 2731. https://doi.org/10.3390/cells12232731
Trstenjak-Prebanda M, Biasizzo M, Dolinar K, Pirkmajer S, Turk B, Brault V, Herault Y, Kopitar-Jerala N. Stefin B Inhibits NLRP3 Inflammasome Activation via AMPK/mTOR Signalling. Cells. 2023; 12(23):2731. https://doi.org/10.3390/cells12232731
Chicago/Turabian StyleTrstenjak-Prebanda, Mojca, Monika Biasizzo, Klemen Dolinar, Sergej Pirkmajer, Boris Turk, Veronique Brault, Yann Herault, and Nataša Kopitar-Jerala. 2023. "Stefin B Inhibits NLRP3 Inflammasome Activation via AMPK/mTOR Signalling" Cells 12, no. 23: 2731. https://doi.org/10.3390/cells12232731