
Levels of Cell-Free DNA in Kidney Failure Patients before and after Renal Transplantation
 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population for Sample Collection
2.2. Measurement of Calcification and Fibrosis Score
2.3. Cell-Free DNA Isolation and Quantification
2.4. Statistical Analysis
3. Results
3.1. Clinical Profile of Study Participants
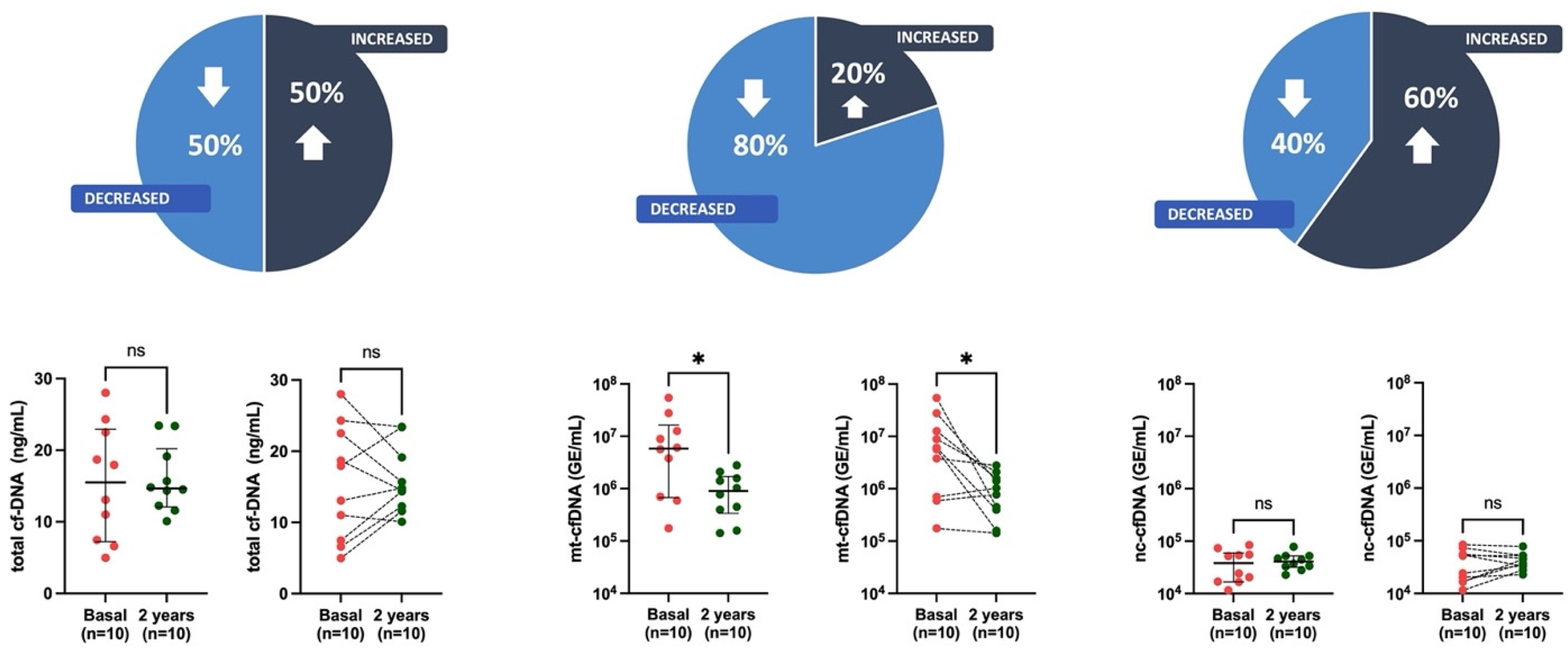
3.2. cfDNA Levels
3.3. cfDNA Levels by Therapy Modality before KT
3.4. cfDNA Levels According to Arterial Calcification and Fibrosis Status
3.5. Sex Differences in cfDNA Levels
3.6. Correlation Analysis
4. Discussion
5. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levey, A.S.; Eckardt, K.U.; Dorman, N.M.; Christiansen, S.L.; Hoorn, E.J.; Ingelfinger, J.R.; Inker, L.A.; Levin, A.; Mehrotra, R.; Palevsky, P.M.; et al. Nomenclature for kidney function and disease: Report of a kidney disease: Improving global outcomes (kdigo) consensus conference. Kidney Int. 2020, 97, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of ckd. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Copur, S.; Tanriover, C.; Yavuz, F.; Galassi, A.; Ciceri, P.; Cozzolino, M. The pathophysiology and management of vascular calcification in chronic kidney disease patients. Expert Rev. Cardiovasc. Ther. 2023, 21, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Podestà, M.A.; Cucchiari, D.; Ciceri, P.; Messa, P.; Torregrosa, J.-V.; Cozzolino, M. Cardiovascular calcifications in kidney transplant recipients. Nephrol. Dial. Transplant. 2022, 37, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in ckd. J. Am. Soc. Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; Alsalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 505, 108–118. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in ckd-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef]
- Ebert, T.; Pawelzik, S.C.; Witasp, A.; Arefin, S.; Hobson, S.; Kublickiene, K.; Shiels, P.G.; Bäck, M.; Stenvinkel, P. Inflammation and premature ageing in chronic kidney disease. Toxins 2020, 12, 227. [Google Scholar] [CrossRef]
- Hobson, S.; Arefin, S.; Kublickiene, K.; Shiels, P.G.; Stenvinkel, P. Senescent cells in early vascular ageing and bone disease of chronic kidney disease-a novel target for treatment. Toxins 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.; Zhao, J.; Contente-Cuomo, T.; Stephens, M.D.; Raupach, E.; Odenheimer-Bergman, A.; Connor, S.; McDonald, B.R.; Moore, B.; Hutchins, E.; et al. Analysis of recurrently protected genomic regions in cell-free dna found in urine. Sci. Transl. Med. 2021, 13, eaaz3088. [Google Scholar] [CrossRef] [PubMed]
- Yuwono, N.L.; Warton, K.; Ford, C.E. The influence of biological and lifestyle factors on circulating cell-free dna in blood plasma. Elife 2021, 10, e69679. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A. Circulating cell-free mitochondrial dna as biomarker of cardiovascular risk: New challenges of old findings. Angiology 2015, 3, 161–164. [Google Scholar] [CrossRef]
- Rosa, H.S.; Ajaz, S.; Gnudi, L.; Malik, A.N. A case for measuring both cellular and cell-free mitochondrial dna as a disease biomarker in human blood. FASEB J. 2020, 34, 12278–12288. [Google Scholar] [CrossRef] [PubMed]
- Trumpff, C.; Michelson, J.; Lagranha, C.J.; Taleon, V.; Karan, K.R.; Sturm, G.; Lindqvist, D.; Fernström, J.; Moser, D.; Kaufman, B.A.; et al. Stress and circulating cell-free mitochondrial dna: A systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion 2021, 59, 225–245. [Google Scholar] [CrossRef]
- Chopra, B.; Sureshkumar, K.K. Emerging role of cell-free dna in kidney transplantation. World J. Exp. Med. 2021, 11, 55–65. [Google Scholar] [CrossRef]
- Jackson, A.M.; Amato-Menker, C.; Bettinotti, M. Cell-free dna diagnostics in transplantation utilizing next generation sequencing. Hum. Immunol. 2021, 82, 850–858. [Google Scholar] [CrossRef]
- Erlandsson, H.; Qureshi, A.R.; Ripsweden, J.; Löfman, I.H.; Söderberg, M.; Wennberg, L.; Lundgren, T.; Bruchfeld, A.; Brismar, T.B.; Stenvinkel, P. Scoring of medial arterial calcification predicts cardiovascular events and mortality after kidney transplantation. J. Intern. Med. 2022, 291, 813–823. [Google Scholar] [CrossRef]
- Hobson, S.; de Loor, H.; Kublickiene, K.; Beige, J.; Evenepoel, P.; Stenvinkel, P.; Ebert, T. Lipid profile is negatively associated with uremic toxins in patients with kidney failure-a tri-national cohort. Toxins 2022, 14, 412. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Larsson, T.E. Chronic kidney disease: A clinical model of premature aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Ward, L.J.; Arefin, S.; Ebert, T.; Laucyte-Cibulskiene, A.; Pilote, L.; Norris, C.M.; Raparelli, V.; Kautzky-Willer, A.; Herrero, M.T.; et al. Blood-brain barrier and gut barrier dysfunction in chronic kidney disease with a focus on circulating biomarkers and tight junction proteins. Sci. Rep. 2022, 12, 4414. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.C.; Sun, J.; Qureshi, A.R.; Dai, L.; Snaedal, S.; Bárány, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P. The higher mortality associated with low serum albumin is dependent on systemic inflammation in end-stage kidney disease. PLoS ONE 2018, 13, e0190410. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Mukai, H.; Lindholm, B.; Heimbürger, O.; Barany, P.; Stenvinkel, P.; Qureshi, A.R. Clinical global assessment of nutritional status as predictor of mortality in chronic kidney disease patients. PLoS ONE 2017, 12, e0186659. [Google Scholar] [CrossRef] [PubMed]
- Snaedal, S.; Qureshi, A.R.; Lund, S.H.; Germanis, G.; Hylander, B.; Heimbürger, O.; Carrero, J.J.; Stenvinkel, P.; Bárány, P. Dialysis modality and nutritional status are associated with variability of inflammatory markers. Nephrol. Dial. Transplant. 2016, 31, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimbürger, O.; Bárány, P.; Lindholm, B.; Lindström, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of cardiovascular disease and mortality risk in patients with advanced ckd. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Janovičová, Ľ.; Konečná, B.; Vlková, B.; Celec, P. Isolation and quantification of extracellular dna from biofluids. Bio-Protocol 2020, 10, e3726. [Google Scholar] [CrossRef]
- Cañas, L.; Iglesias, E.; Pastor, M.C.; Barallat, J.; Juega, J.; Bancu, I.; Lauzurica, R. Inflammation and oxidation: Do they improve after kidney transplantation? Relationship with mortality after transplantation. Int. Urol. Nephrol. 2017, 49, 533–540. [Google Scholar] [CrossRef]
- Coimbra, S.; Rocha, S.; Nascimento, H.; Valente, M.J.; Catarino, C.; Rocha-Pereira, P.; Sameiro-Faria, M.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; et al. Cell-free dna as a marker for the outcome of end-stage renal disease patients on haemodialysis. Clin. Kidney J. 2021, 14, 1371–1378. [Google Scholar] [CrossRef]
- Kloss, K.; Ismail, S.; Redeker, S.; van Hoogdalem, L.; Luchtenburg, A.; Busschbach, J.J.V.; van de Wetering, J. Factors influencing access to kidney transplantation: A research protocol of a qualitative study on stakeholders’ perspectives. BMJ Open 2019, 9, e032694. [Google Scholar] [CrossRef]
- Soeters, P.B.; Wolfe, R.R.; Shenkin, A. Hypoalbuminemia: Pathogenesis and clinical significance. JPEN J. Parenter. Enter. Nutr. 2019, 43, 181–193. [Google Scholar] [CrossRef]
- Kim, K.; Moon, H.; Lee, Y.H.; Seo, J.-W.; Kim, Y.G.; Moon, J.-Y.; Kim, J.S.; Jeong, K.-H.; Lee, T.W.; Ihm, C.-G.; et al. Clinical relevance of cell-free mitochondrial dna during the early postoperative period in kidney transplant recipients. Sci. Rep. 2019, 9, 18607. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early vascular ageing and cellular senescence in chronic kidney disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef]
- Hobson, S.; Arefin, S.; Witasp, A.; Hernandez, L.; Kublickiene, K.; Shiels, P.; Stenvinkel, P. Accelerated vascular aging in chronic kidney disease: The potential for novel therapies. Circ. Res. 2023, 132, 950–969. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Sonntag, W.E.; Csiszar, A. Mitochondria and aging in the vascular system. J. Mol. Med. 2010, 88, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. From mitochondria to atherosclerosis: The inflammation path. Biomedicines 2021, 9, 258. [Google Scholar] [CrossRef] [PubMed]
- Tantisattamo, E.; Ho, B.T.; Workeneh, B.T. Editorial: Metabolic changes after kidney transplantation. Front. Med. 2021, 8, 709644. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Moroni, G.; Ponticelli, C. Cardiovascular risk after kidney transplantation: Causes and current approaches to a relevant burden. J. Pers. Med. 2022, 12, 1200. [Google Scholar] [CrossRef] [PubMed]
- Nemati, E.; Einollahi, B.; Pezeshki, M.L.; Porfarziani, V.; Fattahi, M.R. Does kidney transplantation with deceased or living donor affect graft survival? Nephro-Urol. Mon. 2014, 6, e12182. [Google Scholar] [CrossRef]
- Mix, T.C.H.; Kazmi, W.; Khan, S.; Ruthazer, R.; Rohrer, R.; Pereira, B.J.G.; Kausz, A.T. Anemia: A continuing problem following kidney transplantation. Am. J. Transplant. 2003, 3, 1426–1433. [Google Scholar] [CrossRef]
- Schechter, A.; Gafter-Gvili, A.; Shepshelovich, D.; Rahamimov, R.; Gafter, U.; Mor, E.; Chagnac, A.; Rozen-Zvi, B. Post renal transplant anemia: Severity, causes and their association with graft and patient survival. BMC Nephrol. 2019, 20, 51. [Google Scholar] [CrossRef]
- Agrawal, A.; Ison, M.G.; Danziger-Isakov, L. Long-term infectious complications of kidney transplantation. Clin. J. Am. Soc. Nephrol. 2022, 17, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Gioco, R.; Corona, D.; Ekser, B.; Puzzo, L.; Inserra, G.; Pinto, F.; Schipa, C.; Privitera, F.; Veroux, P.; Veroux, M. Gastrointestinal complications after kidney transplantation. World J. Gastroenterol. 2020, 26, 5797–5811. [Google Scholar] [CrossRef] [PubMed]
- Ceprian, N.; Valera, G.; Caro, J.; Yuste, C.; Serroukh, N.; de Pablos, I.G.; Oliva, C.; Figuer, A.; Praga, M.; Alique, M.; et al. Effect of kidney transplantation on accelerated immunosenescence and vascular changes induced by chronic kidney disease. Front. Med. 2021, 8, 705159. [Google Scholar] [CrossRef]
- Bellini, M.I.; Deurloo, E.; Consorti, F.; Herbert, P.E. Body mass index affects kidney transplant outcomes: A cohort study over 5 years using a steroid sparing protocol. Front. Endocrinol. 2023, 14, 1106087. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Miyata, H.; Aikou, S.; Seto, Y.; Akazawa, K.; Takeuchi, M.; Matsui, Y.; Konno, H.; Gotoh, M.; Mori, M.; et al. Effects of body mass index (bmi) on surgical outcomes: A nationwide survey using a japanese web-based database. Surg. Today 2015, 45, 1271–1279. [Google Scholar] [CrossRef]
- Ammar, A. The role of ketoacid analogs with dietary protein restriction in chronic kidney disease. Int. J. Res. Publ. 2022, 93, 178–188. [Google Scholar] [CrossRef]
- Oh, S.E.; Lee, Y.-K.; Kim, J.K.; Lee, D.H.; Kim, S.-J.; Kim, S.G.; Oh, J.E.; Seo, J.W.; Yoon, J.-W.; Koo, J.-R.; et al. Effects of keto acids on serum albumin level in hemodialysis patients with hypoalbuminemia. Korean J. Nephrol. 2010, 29, 82–88. Available online: https://www.krcp-ksn.org/journal/view.php?number=5864 (accessed on 21 November 2023).
- Bissoli, L.; Di Francesco, V.; Ballarin, A.; Mandragona, R.; Trespidi, R.; Brocco, G.; Caruso, B.; Bosello, O.; Zamboni, M. Effect of vegetarian diet on homocysteine levels. Ann. Nutr. Metab. 2002, 46, 73–79. [Google Scholar] [CrossRef]
- Badri, S.; Vahdat, S.; Seirafian, S.; Pourfarzam, M.; Gholipur-Shahraki, T.; Ataei, S. Homocysteine-lowering interventions in chronic kidney disease. J. Res. Pharm. Pract. 2021, 10, 114–124. [Google Scholar] [CrossRef]
- Abeysekera, R.A.; Healy, H.G.; Wang, Z.; Cameron, A.L.; Hoy, W.E. Heterogeneity in patterns of progression of chronic kidney disease. Intern. Med. J. 2021, 51, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kang, A.; Nigwekar, S.U.; Perkovic, V.; Kulshrestha, S.; Zoungas, S.; Navaneethan, S.D.; Cass, A.; Gallagher, M.P.; Ninomiya, T.; Strippoli, G.F.; et al. Interventions for lowering plasma homocysteine levels in kidney transplant recipients. Cochrane Database Syst. Rev. 2015, 5, CD007910. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, M.; Ayis, S.; Tuddenham, E.; Lo, J.; Lei, K.; Smith, J.; Sanderson, B.; Moran, C.; Collinson, P.; Peacock, J.; et al. Cardiac troponin release is associated with biomarkers of inflammation and ventricular dilatation during critical illness. Shock 2017, 47, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Tovbin, D.; Novack, V.; Wiessman, M.P.; Elkadir, A.A.; Zlotnik, M.; Douvdevani, A. Circulating cell-free dna in hemodialysis patients predicts mortality. Nephrol. Dial. Transplant. 2012, 27, 3929–3935. [Google Scholar] [CrossRef]
- Filippone, E.J.; Farber, J.L. The monitoring of donor-derived cell-free dna in kidney transplantation. Transplantation 2021, 105, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Ducloux, D.; Legendre, M.; Bamoulid, J.; Rebibou, J.-M.; Saas, P.; Courivaud, C.; Crepin, T. Esrd-associated immune phenotype depends on dialysis modality and iron status: Clinical implications. Immun. Ageing 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Mikael, L.D.R.; De Paiva, A.M.G.; Gomes, M.M.; Sousa, A.L.L.; Jardim, P.C.B.V.; Vitorino, P.V.D.O.; Euzébio, M.B.; Sousa, W.D.M.; Barroso, W.K.S. Vascular aging and arterial stiffness. Arq. Bras. Cardiol. 2017, 109, 253–258. [Google Scholar] [CrossRef]
- Bitenc, M.; Tune, B.G.; Melheim, M.; Atneosen-Åsegg, M.; Lai, X.; Rajar, P.; Solberg, R.; Baumbusch, L.O. Assessing nuclear versus mitochondrial cell-free dna (cfdna) by qrt-pcr and droplet digital pcr using a piglet model of perinatal asphyxia. Mol. Biol. Rep. 2023, 50, 1533–1544. [Google Scholar] [CrossRef]
- London, G.M.; Drüeke, T.B. Atherosclerosis and arteriosclerosis in chronic renal failure. Kidney Int. 1997, 51, 1678–1695. [Google Scholar] [CrossRef]
- Park, K.A.; Jo, H.M.; Han, J.S.; Kim, M.J.; Kwun, D.H.; Park, M.Y.; Choi, S.J.; Kim, J.K.; Hwang, S.D. Features of atherosclerosis in hemodialysis patients. Kidney Res. Clin. Pract. 2013, 32, 177–182. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular fibrosis in aging and hypertension: Molecular mechanisms and clinical implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in chronic kidney disease: Pathogenesis and consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Atherosclerosis specific features in chronic kidney disease (ckd). Biomedicines 2022, 10, 2094. [Google Scholar] [CrossRef]
- Meddeb, R.; Dache, Z.A.A.; Thezenas, S.; Otandault, A.; Tanos, R.; Pastor, B.; Sanchez, C.; Azzi, J.; Tousch, G.; Azan, S.; et al. Quantifying circulating cell-free dna in humans. Sci. Rep. 2019, 9, 5220. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Lu, J.; Wang, L.; Gao, M. Pro-inflammatory role of cell-free mitochondrial dna in cardiovascular diseases. IUBMB Life 2020, 72, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Alghofaili, L.; Almubarak, H.; Gassem, K.; Islam, S.S.; Coskun, S.; Kaya, N.; Karakas, B. Cell-free dna levels of twins and sibling pairs indicate individuality and possible use as a personalized biomarker. PLoS ONE 2019, 14, e0223470. [Google Scholar] [CrossRef] [PubMed]
- Kananen, L.; Hurme, M.; Bürkle, A.; Moreno-Villanueva, M.; Bernhardt, J.; Debacq-Chainiaux, F.; Grubeck-Loebenstein, B.; Malavolta, M.; Basso, A.; Piacenza, F.; et al. Circulating cell-free dna in health and disease-the relationship to health behaviours, ageing phenotypes and metabolomics. Geroscience 2023, 45, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Hyppönen, E. Vitamin d deficiency and c-reactive protein: A bidirectional mendelian randomization study. Int. J. Epidemiol. 2023, 52, 260–271. [Google Scholar] [CrossRef]
- Bassiouni, W.; Ali, M.A.M.; Schulz, R. Multifunctional intracellular matrix metalloproteinases: Implications in disease. FEBS J. 2021, 288, 7162–7182. [Google Scholar] [CrossRef]
- Ravindranath, M.H.; El Hilali, F.; Filippone, E.J. The impact of inflammation on the immune responses to transplantation: Tolerance or rejection? Front. Immunol. 2021, 12, 667834. [Google Scholar] [CrossRef]
- Heldal, T.F.; Åsberg, A.; Ueland, T.; Reisæter, A.V.; Pischke, S.E.; Mollnes, T.E.; Aukrust, P.; Hartmann, A.; Heldal, K.; Jenssen, T. Inflammation in the early phase after kidney transplantation is associated with increased long-term all-cause mortality. Am. J. Transplant. 2022, 22, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Lauková, L.; Konečná, B.; Bábíčková, J.; Wagnerová, A.; Melišková, V.; Vlková, B.; Celec, P. Exogenous deoxyribonuclease has a protective effect in a mouse model of sepsis. Biomed. Pharmacother. 2017, 93, 8–16. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basal (n = 44) | Two Years Post KT (n = 44) | p-Value | |
|---|---|---|---|
| Age, years | 45 (32–50) | 47 (34–52) | // |
| BMI, kg/m2 | 23.6 (21.7–28.1), n = 21 | 25.7 (22.9–29.3), n = 21 | 0.0001 * |
| Males | 33 (75) | ||
| SBP, mmHg | 147 (123–167), n = 25 | 138 (125–147), n =25 | 0.3155 |
| DBP, mmHg | 86 (76–97), n = 25 | 78 (75–80), n =25 | 0.1167 |
| CVD | 39 (89) | // | // |
| DM | 3 (7) | // | // |
| Mode of therapy pre-KT (HD) | 22 (50) | // | // |
| Medications at cohort entry | |||
| ACE inhibitors/ARBs | 25 (56) | 28 (64) | 0.6534 |
| Beta-blockers | 30 (32) | 23 (43) | 0.1273 |
| Ca2+ channel blockers | 26 (59) | 15 (34) | 0.0187 * |
| Statins | 9 (26) | 29 (66) | 0.0005 * |
| Laboratory Data | |||
| Cholesterol, mmol/L | 4.5 (3.8–5.0) | 4.2 (3.9–4.9), n = 39 | 0.9685 |
| HDL, mmol/L | 1.4 (1.1–1.6), n = 39 | 1.5 (1.1–1.7), n = 39, n = 39 | 0.0388 * |
| Triglycerides, mmol/L | 1.3 (1.0–1.8), n = 38 | 1.4 (1.0–2.0), n = 38 | 0.4991 |
| Lp (a), mg/L | 49 (13–102), n = 27 | 12 (10–73), n = 27 | 0.0112 * |
| Apo-A1, g/L | 1.4 (1.2–1.6), n = 39 | 1.5 (1.3–1.8), n = 39 | 0.0007 * |
| Apo-B, g/L | 0.9 (0.7–1.0), n = 39 | 0.7 (0.6–1.0), n = 39 | 0.1858 |
| Creatinine, mg/dL | 7.3 (6.4–9.8) | 1.2 (1.1–1.4) | <0.0001 * |
| Albumin, g/L | 36 (33–39), n = 42 | 38 (36–40), n = 42 | 0.0070 * |
| HbA1c, % | 5.3 (4.9–5.5), n = 38 | 5.5 (5.4–6.0), n = 38 | <0.0001 * |
| hs-CRP, mg/L | 1.0 (0.3–2.5), n = 43 | 0.9 (0.5–2.4), n = 43 | 0.9581 |
| 25 (OH) vitD, nmol/L | 45. (35–66), n = 39 | 57 (39–82), n = 39 | 0.1601 |
| Homocysteine, µmol/L | 36 (28.0–48.5), n = 40 | 18 (14- 21), n = 40 | <0.0001 * |
| Calcium, mmol/L | 2.2 (2.1–2.4), n = 41 | 2.4 (2.3–2.4), n = 41 | <0.0001 * |
| P-Phosphate, mmol/L | 1.7 (1.2–1.9), n = 41 | 1.0 (0.8–1.0), n = 41 | <0.0001 * |
| Folate, nmol/L | 12 (8–38), n = 42 | 13 (11–17), n = 42 | 0.6153 |
| P-Troponin T, µg/L | 20.0 (14.0–36.3), n = 38 | 8.0 (6.0–11.3), n =38 | <0.0001 * |
| Calprotectin, µg/ml | 2.3 (1.7–3.2), n = 38 | 1.6 (1.0–2.9), n = 38 | 0.3280 |
| MMP-9, ng/mL | 380 (290–579), n = 34 | // | // |
| Increased Trend | Decreased Trend | p-Value | |
|---|---|---|---|
| Total cfDNA | n = 20 | n = 24 | |
| BMI, kg/m2 | 24.4 (22.3–29.4) | 22.4 (20.4–26.1) | 0.0388 * |
| Albumin, g/L | 38 (36–40), n = 19 | 34 (31.3–36.8) | 0.0015 * |
| Homocysteine, µmol/L | 32.5 (26.5–39.8) | 38.5 (32–69.3) | 0.0295 * |
| P-troponin T, µg/L | 24.0 (17.5–48.5), n = 19 | 17.5 (12.8–23.8) | 0.0371 * |
| mt-cfDNA | n = 15 | n = 29 | |
| -- | -- | -- | -- |
| nc-cfDNA | n = 23 | n = 21 | |
| Albumin, g/L | 37 (35–40) | 33 (30–38), n = 20 | 0.0043 * |
| SBP, mmHg | 132 (127–142) | 153 (128–172) | 0.0406 * |
| Basal (n = 44) | 2 Years Post-KT (n = 44) | |||
|---|---|---|---|---|
| R | p Value | r | p-Value | |
| total cfDNA | ||||
| SBP, mmHg | 0.312 | 0.040 * | 0.036, n = 25 | 0.7341 |
| hs-CRP, mg/L | 0.029 | 0.852 | 0.322, n = 43 | 0.035 * |
| MMP-9, ng/mL | −0.383, n = 34 | 0.025 * | -- | -- |
| Vintage, years | 0.527, n = 28 | 0.040 * | -- | -- |
| mt-cfDNA | ||||
| hsCRP, mg/L | −0.141 | 0.360 | 0.309, n = 43 | 0.044 * |
| Apo-B, g/L | 0.071 | 0.649 | 0.333, n = 39 | 0.039 * |
| Homocysteine, µmol/L | 0.399 | 0.007 * | 0.107. n = 40 | 0.510 |
| Lp (a), mg/L | 0.514, n = 32 | 0.003 * | 0.324, n = 39 | 0.044 * |
| 25-(OH) Vit-D, nmol/L | −0.181 | 0.240 | −0.333, n = 39 | 0.038 * |
| nc-cfDNA | ||||
| SBP, mmHg | 0.312 | 0.039 * | −0.005, n = 25 | 0.9810 |
| Calprotectin, µg/ml | 0.357, n = 39 | 0.026 * | 0.184, n = 38 | 0.269 |
| MMP9, ng/mL | −0.349, n = 34 | 0.043 * | -- | -- |
| Vintage, years | 0.489, n = 28 | 0.008 * | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leotta, C.; Hernandez, L.; Tothova, L.; Arefin, S.; Ciceri, P.; Cozzolino, M.G.; Barany, P.; Chromek, M.; Stenvinkel, P.; Kublickiene, K. Levels of Cell-Free DNA in Kidney Failure Patients before and after Renal Transplantation. Cells 2023, 12, 2774. https://doi.org/10.3390/cells12242774
Leotta C, Hernandez L, Tothova L, Arefin S, Ciceri P, Cozzolino MG, Barany P, Chromek M, Stenvinkel P, Kublickiene K. Levels of Cell-Free DNA in Kidney Failure Patients before and after Renal Transplantation. Cells. 2023; 12(24):2774. https://doi.org/10.3390/cells12242774
Chicago/Turabian StyleLeotta, Chiara, Leah Hernandez, Lubomira Tothova, Samsul Arefin, Paola Ciceri, Mario Gennaro Cozzolino, Peter Barany, Milan Chromek, Peter Stenvinkel, and Karolina Kublickiene. 2023. "Levels of Cell-Free DNA in Kidney Failure Patients before and after Renal Transplantation" Cells 12, no. 24: 2774. https://doi.org/10.3390/cells12242774