
FoxO3a Drives the Metabolic Reprogramming in Tamoxifen-Resistant Breast Cancer Cells Restoring Tamoxifen Sensitivity

 ,
,  ,
,  ,
,  ,
,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of Tam Resistant Cell Lines
2.2. FoxO3a Inducible Stable Cell Lines
2.3. Plasmids and Transient Transfections
2.4. RNA Extraction, Reverse Transcription, and Real-Time (RT)-PCR
2.5. Western Blotting (WB)
2.6. Glucose Assay
2.7. Lactate Dehydrogenase (LDH) Activity Assay
2.8. Glucose-6-Phosphate Dehydrogenase (G6PDH) Activity Assay
2.9. Seahorse XFe96 Metabolic Flux Analysis
2.10. Sulfo-Rhodamine B (SRB) Assay
2.11. Label-Free Unbiased Semi-Quantitative Proteomics Analysis
2.12. Mitochondrial Staining
2.13. Reactive Oxygen Species (ROS) Assessment
2.14. cBioPortal Analysis
2.15. Statistical Analysis
3. Results
3.1. Nuclear FoxO3a Restores the Sensitivity of TamR BCCs to Tam
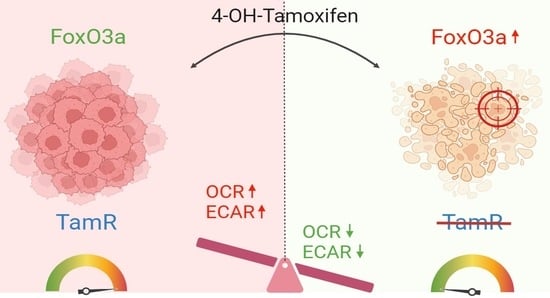
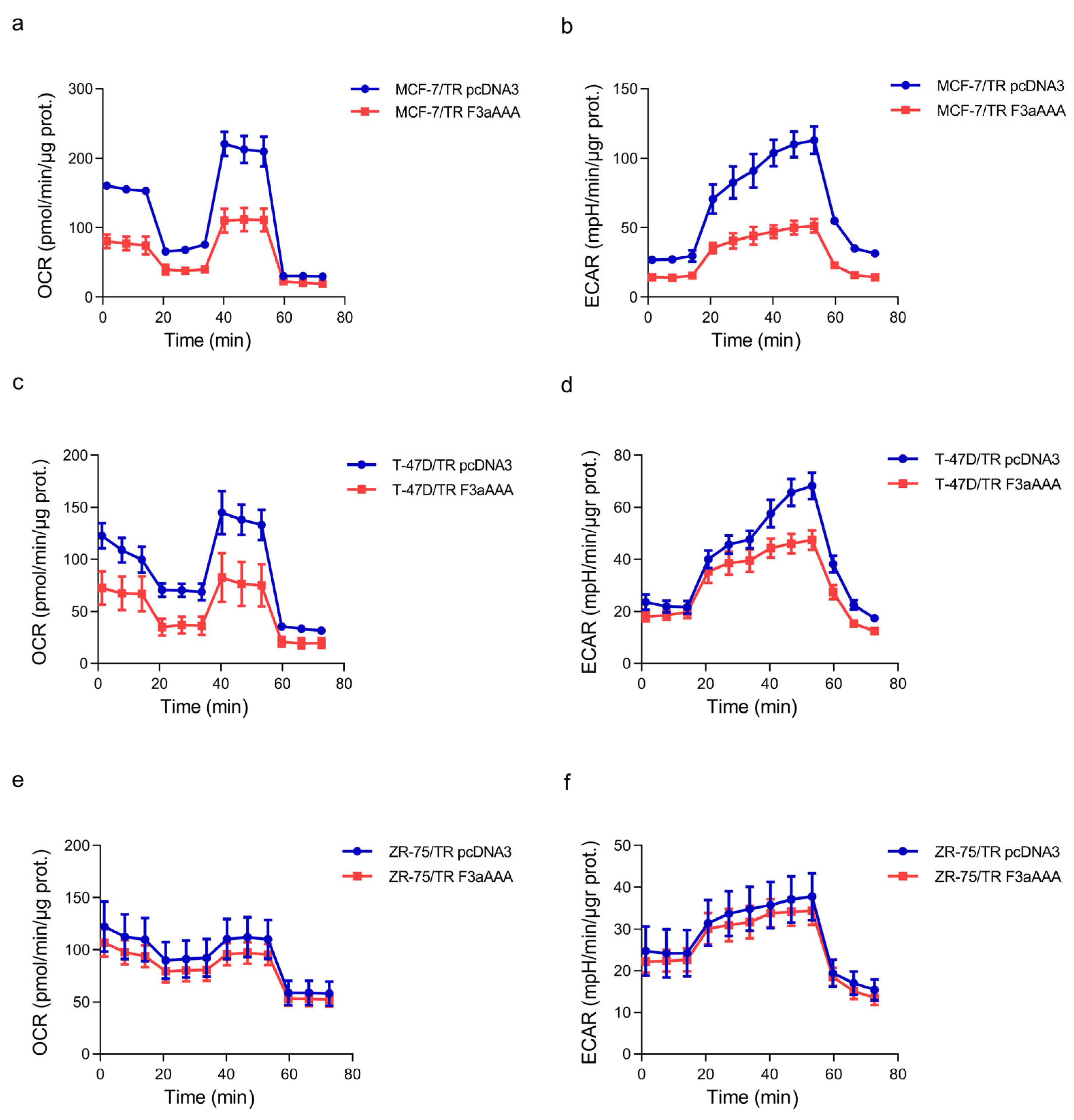
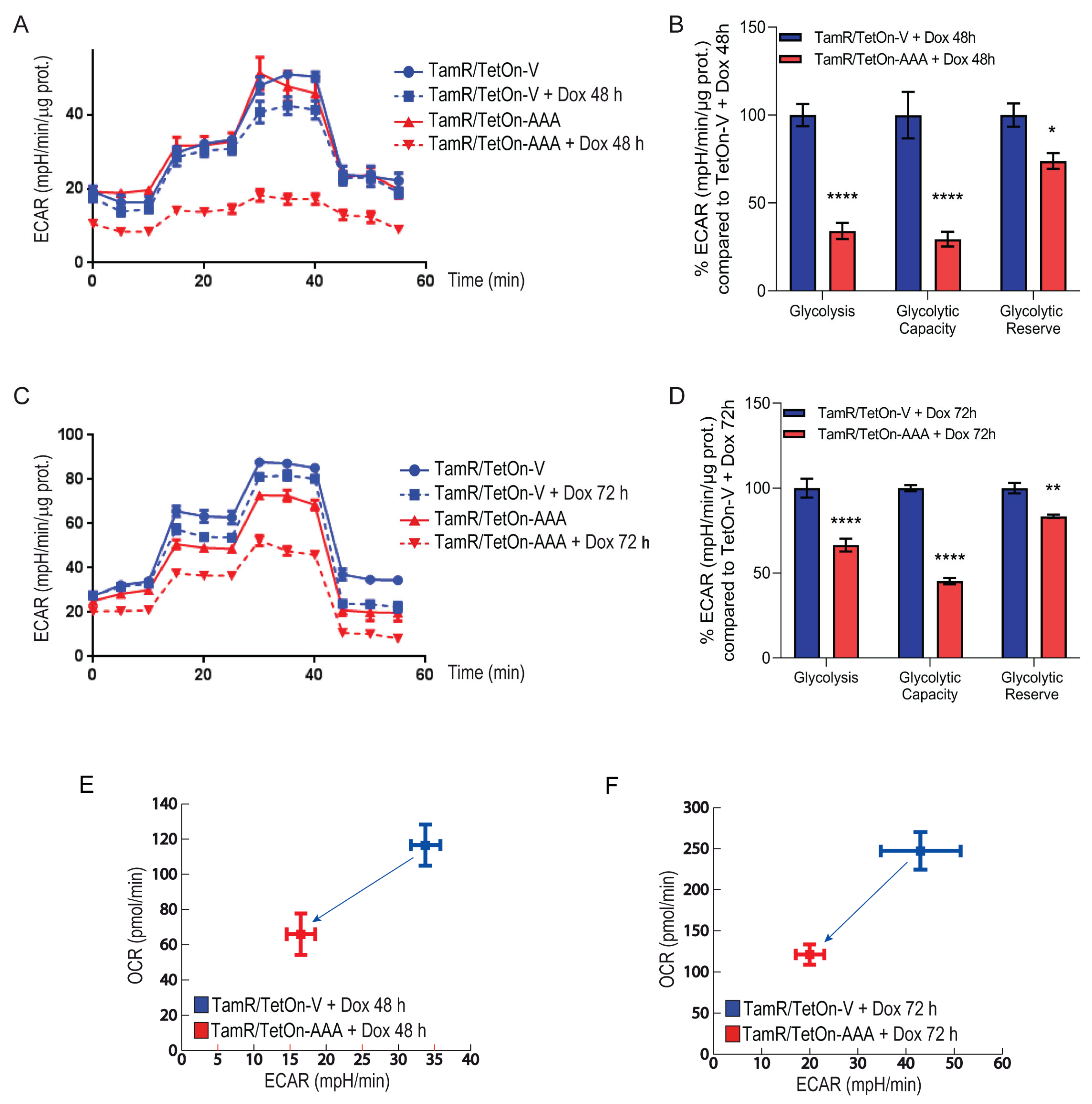
3.2. FoxO3a Counteracts the Increased Oxygen Consumption Rate and Extracellular Acidification Rate Observed in TamR BCCs
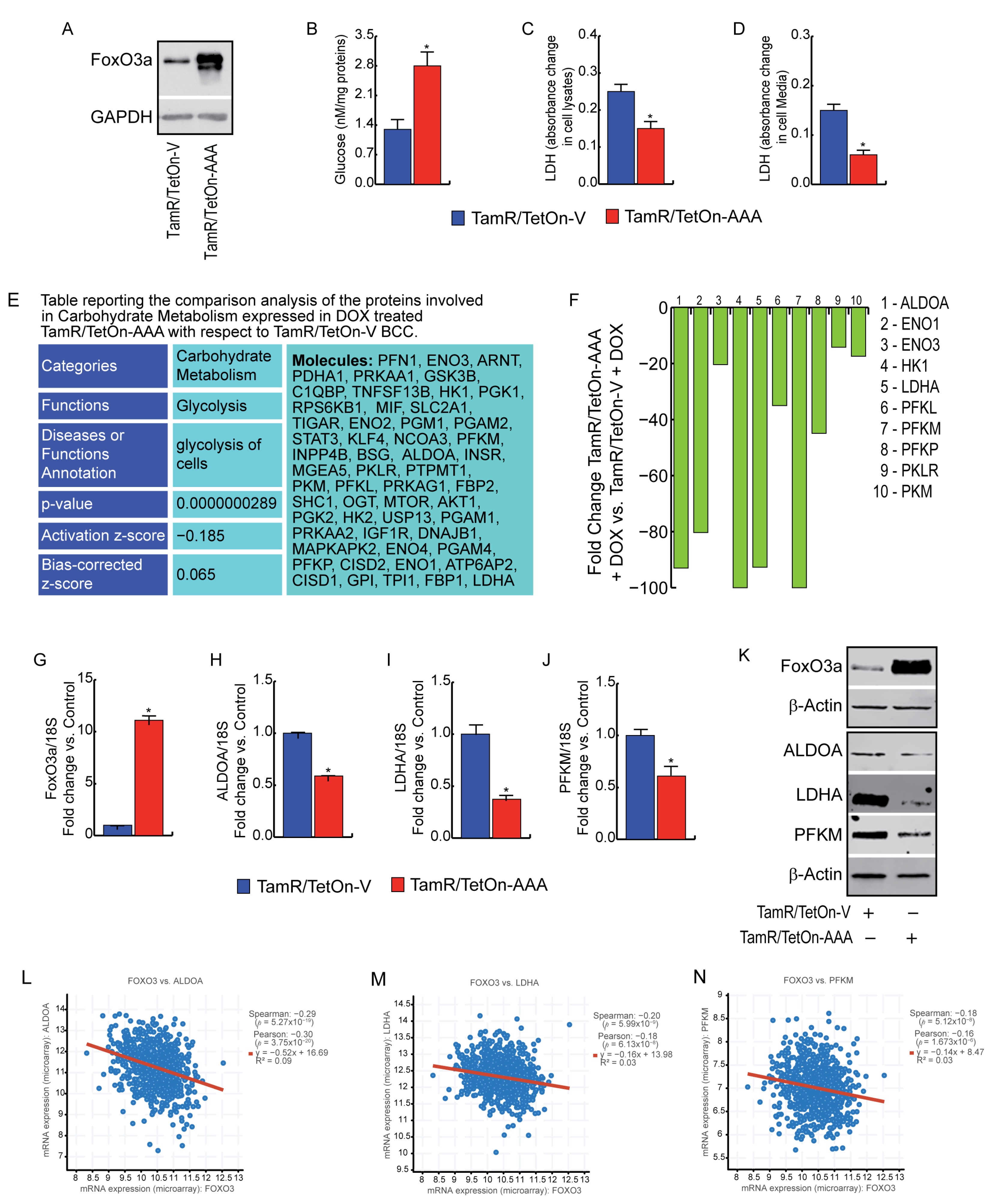
3.3. FoxO3a Reduces Glycolysis Efficiency and LDH Activity in TamR BCCs
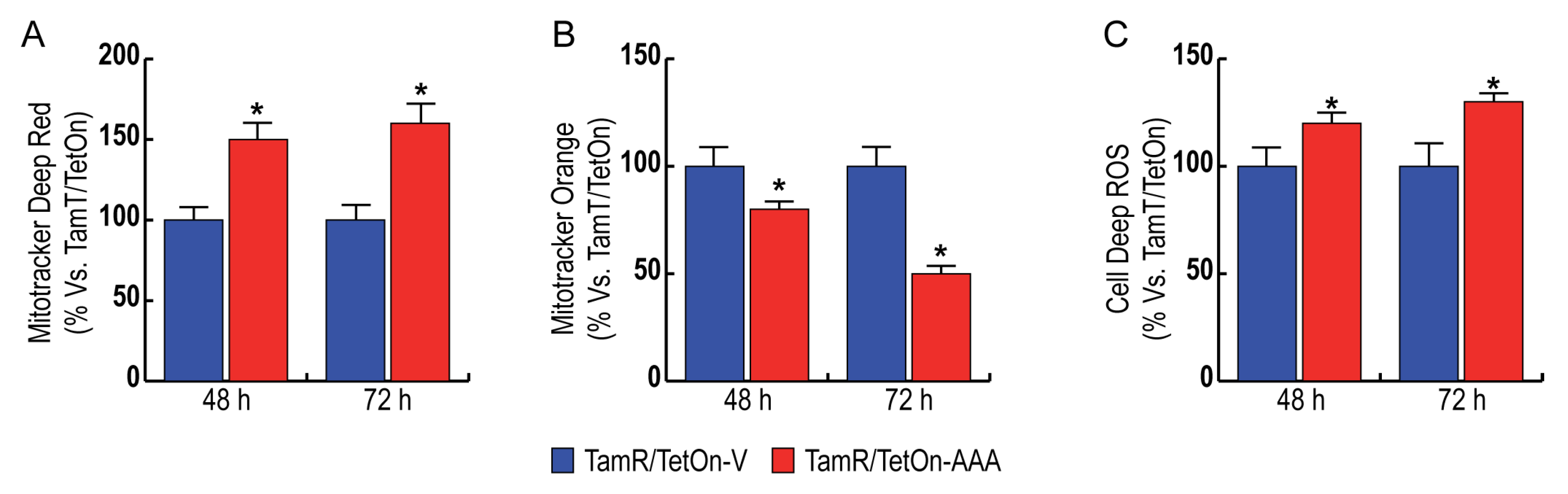
3.4. FoxO3a Increases Mitochondrial Biogenesis and Reduces Mitochondrial Functionality, Increasing ROS Production in TamR BCCs
3.5. FoxO3a Impairs NADPH Production through the Pentose Phosphate Pathway (PPP) in TamR BCCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Kim, J.S.; Jiang, J.; Peston, D.; Sinnett, H.D.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr. -Relat. Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sotgia, F.; Sisci, D.; Cappello, A.R.; Lisanti, M.P. Mitochondrial “power” drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget 2017, 8, 20309–20327. [Google Scholar] [CrossRef]
- Harrelson, J.P.; Lee, M.W. Expanding the view of breast cancer metabolism: Promising molecular targets and therapeutic opportunities. Pharmacol. Ther. 2016, 167, 60–73. [Google Scholar] [CrossRef]
- Lu, X.; Bennet, B.; Mu, E.; Rabinowitz, J.; Kang, Y. Metabolomic changes accompanying transformation and acquisition of metastatic potential in a syngeneic mouse mammary tumor model. J. Biol. Chem. 2010, 285, 9317–9321. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Khatri, S.; Yepiskoposyan, H.; Gallo, C.A.; Tandon, P.; Plas, D.R. FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J. Biol. Chem. 2010, 285, 15960–15965. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Chiacchiera, F.; Grossi, V.; Matrone, A.; Latorre, D.; Simonatto, M.; Fusella, A.; Ryall, J.G.; Finley, L.W.; Haigis, M.C.; et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell. Mol. Life Sci. CMLS 2013, 70, 2015–2029. [Google Scholar] [CrossRef] [PubMed]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Li, X.; Jia, Y.; Wang, J.; Ao, X. FOXO3a in cancer drug resistance. Cancer Lett. 2022, 540, 215724. [Google Scholar] [CrossRef]
- Pellegrino, M.; Rizza, P.; Donà, A.; Nigro, A.; Ricci, E.; Fiorillo, M.; Perrotta, I.; Lanzino, M.; Giordano, C.; Bonofiglio, D. FoxO3a as a Positive Prognostic Marker and a Therapeutic Target in Tamoxifen-Resistant Breast Cancer. Cancer 2019, 11, 1858. [Google Scholar] [CrossRef]
- Pellegrino, M.; Rizza, P.; Nigro, A.; Ceraldi, R.; Ricci, E.; Perrotta, I.; Aquila, S.; Lanzino, M.; Andò, S.; Morelli, C.; et al. FoxO3a Mediates the Inhibitory Effects of the Antiepileptic Drug Lamotrigine on Breast Cancer Growth. Mol. Cancer Res. MCR 2018, 16, 923–934. [Google Scholar] [CrossRef]
- Ricci, E.; Fava, M.; Rizza, P.; Pellegrino, M.; Bonofiglio, D. FoxO3a Inhibits Tamoxifen-Resistant Breast Cancer Progression by Inducing Integrin α5 Expression. Cancers 2022, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Kaja, S.; Payne, A.J.; Singh, T.; Ghuman, J.K.; Sieck, E.G.; Koulen, P. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. J. Pharmacol. Toxicol. Methods 2015, 73, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alvarez, R.; De Francesco, E.M.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P. Mitochondrial Fission Factor (MFF) Inhibits Mitochondrial Metabolism and Reduces Breast Cancer Stem Cell (CSC) Activity. Front. Oncol. 2020, 10, 1776. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Frattaruolo, L. New insights into cholesterol-mediated ERRα activation in breast cancer progression and pro-tumoral microenvironment orchestration. FEBS J. 2022, 290, 1481–1501. [Google Scholar] [CrossRef]
- Morelli, C.; Lanzino, M.; Garofalo, C.; Maris, P.; Brunelli, E.; Casaburi, I.; Catalano, S.; Bruno, R.; Sisci, D.; Andò, S. Akt2 inhibition enables the forkhead transcription factor FoxO3a to have a repressive role in estrogen receptor alpha transcriptional activity in breast cancer cells. Mol. Cell. Biol. 2010, 30, 857–870. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The Multiple Roles of Glucose-6-Phosphate Dehydrogenase in Tumorigenesis and Cancer Chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Xu, I.M.-J.; Lai, R.K.-H.; Lin, S.-H.; Tse, A.P.-W.; Chiu, D.K.-C.; Koh, H.-Y.; Law, C.-T.; Wong, C.-M.; Cai, Z.; Wong, C.C.-L.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef]
- Qin, Z.; Xiang, C.; Zhong, F.; Liu, Y.; Dong, Q.; Li, K.; Shi, W.; Ding, C.; Qin, L.; He, F. Transketolase (TKT) activity and nuclear localization promote hepatocellular carcinoma in a metabolic and a non-metabolic manner. J. Exp. Clin. Cancer Res. CR 2019, 38, 154. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, H.; Wu, L.; Lai, Z.; Geng, D.; Yang, W.; Zhang, J.; Fan, Z.; Qin, W.; Wang, Y.; et al. O-GlcNAcylation promotes pancreatic tumor growth by regulating malate dehydrogenase 1. Nat. Chem. Biol. 2022, 18, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Godesi, S.; Han, J.R.; Kim, J.K.; Kwak, D.I.; Lee, J.; Nada, H.; Kim, M.; Yang, H.A.; Im, J.Y.; Ban, H.S.; et al. Design, Synthesis and Biological Evaluation of Novel MDH Inhibitors Targeting Tumor Microenvironment. Pharmaceuticals 2023, 16, 683. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.J.; Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Davie, J.R. An integrated analysis of genes and pathways exhibiting metabolic differences between estrogen receptor positive breast cancer cells. BMC Cancer 2007, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Perren, A.; Weng, L.P.; Boag, A.H.; Ziebold, U.; Thakore, K.; Dahia, P.L.; Komminoth, P.; Lees, J.A.; Mulligan, L.M.; Mutter, G.L.; et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am. J. Pathol. 1999, 155, 1253–1260. [Google Scholar] [CrossRef]
- Jang, K.; Kim, M.; Seo, H.S.; Shin, I. PTEN sensitizes MDA-MB-468 cells to inhibition of MEK/Erk signaling for the blockade of cell proliferation. Oncol. Rep. 2010, 24, 787–793. [Google Scholar] [CrossRef]
- Liu, D.; Wang, D.; Wu, C.; Zhang, L.; Mei, Q.; Hu, G.; Long, G.; Sun, W. Prognostic significance of serum lactate dehydrogenase in patients with breast cancer: A meta-analysis. Cancer Manag. Res. 2019, 11, 3611–3619. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Borniquel, S.; García-Quintáns, N.; Valle, I.; Olmos, Y.; Wild, B.; Martínez-Granero, F.; Soria, E.; Lamas, S.; Monsalve, M. Inactivation of Foxo3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Mol. Cell. Biol. 2010, 30, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z. FoxO transcription factors in mitochondrial homeostasis. Biochem. J. 2022, 479, 525–536. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorillo, M.; Ricci, E.; Fava, M.; Longobucco, C.; Sotgia, F.; Rizza, P.; Lanzino, M.; Bonofiglio, D.; Conforti, F.L.; Catalano, S.; et al. FoxO3a Drives the Metabolic Reprogramming in Tamoxifen-Resistant Breast Cancer Cells Restoring Tamoxifen Sensitivity. Cells 2023, 12, 2777. https://doi.org/10.3390/cells12242777
Fiorillo M, Ricci E, Fava M, Longobucco C, Sotgia F, Rizza P, Lanzino M, Bonofiglio D, Conforti FL, Catalano S, et al. FoxO3a Drives the Metabolic Reprogramming in Tamoxifen-Resistant Breast Cancer Cells Restoring Tamoxifen Sensitivity. Cells. 2023; 12(24):2777. https://doi.org/10.3390/cells12242777
Chicago/Turabian StyleFiorillo, Marco, Elena Ricci, Mariarosa Fava, Camilla Longobucco, Federica Sotgia, Pietro Rizza, Marilena Lanzino, Daniela Bonofiglio, Francesca Luisa Conforti, Stefania Catalano, and et al. 2023. "FoxO3a Drives the Metabolic Reprogramming in Tamoxifen-Resistant Breast Cancer Cells Restoring Tamoxifen Sensitivity" Cells 12, no. 24: 2777. https://doi.org/10.3390/cells12242777
APA StyleFiorillo, M., Ricci, E., Fava, M., Longobucco, C., Sotgia, F., Rizza, P., Lanzino, M., Bonofiglio, D., Conforti, F. L., Catalano, S., Barone, I., Morelli, C., Aquila, S., Lisanti, M. P., & Sisci, D. (2023). FoxO3a Drives the Metabolic Reprogramming in Tamoxifen-Resistant Breast Cancer Cells Restoring Tamoxifen Sensitivity. Cells, 12(24), 2777. https://doi.org/10.3390/cells12242777