
A Mouse Model to Study the Pathogenesis of γ-herpesviral Infections in Germinal Center B Cells

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mice
2.2. BAC Mutagenesis of MHV-68 hNGFR and MHV-68 LMP2A
2.3. MHV-68 Infection and NP-CGG Immunization
2.4. Organ Preparation and the Determination of Cell Numbers
2.5. Flow Cytometry
2.6. The Detection of Virus-Positive Cells Using the PrimeFlow RNA Assay
2.7. The Measurement of Viral Genomic Load by Using Quantitative Real-Time PCR
2.8. Statistics
3. Results
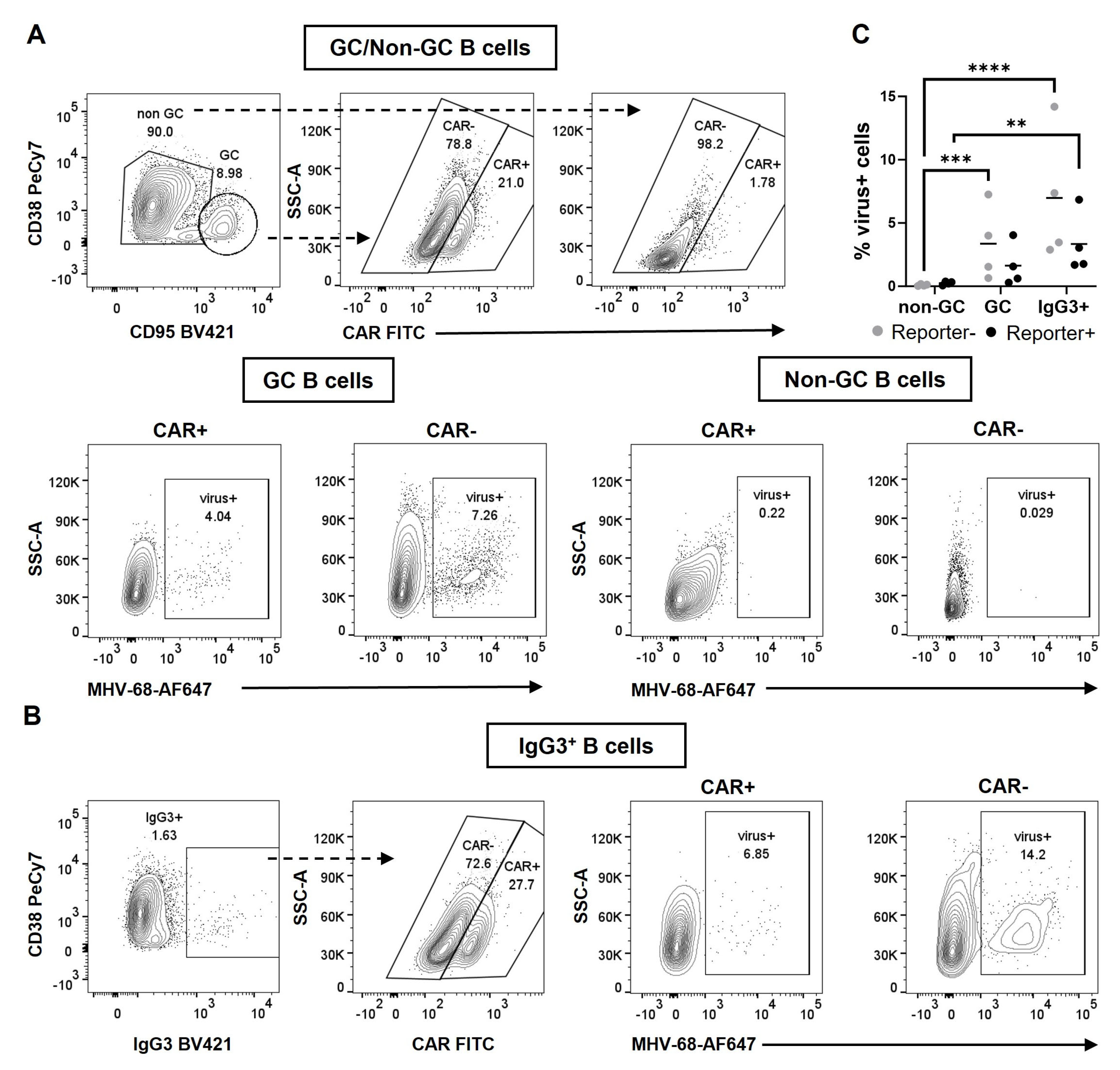
3.1. MHV-68 hNGFR Infection of CAR//Cγ1-cre Reporter Mice Results in Cre-Mediated Recombination in B Cells
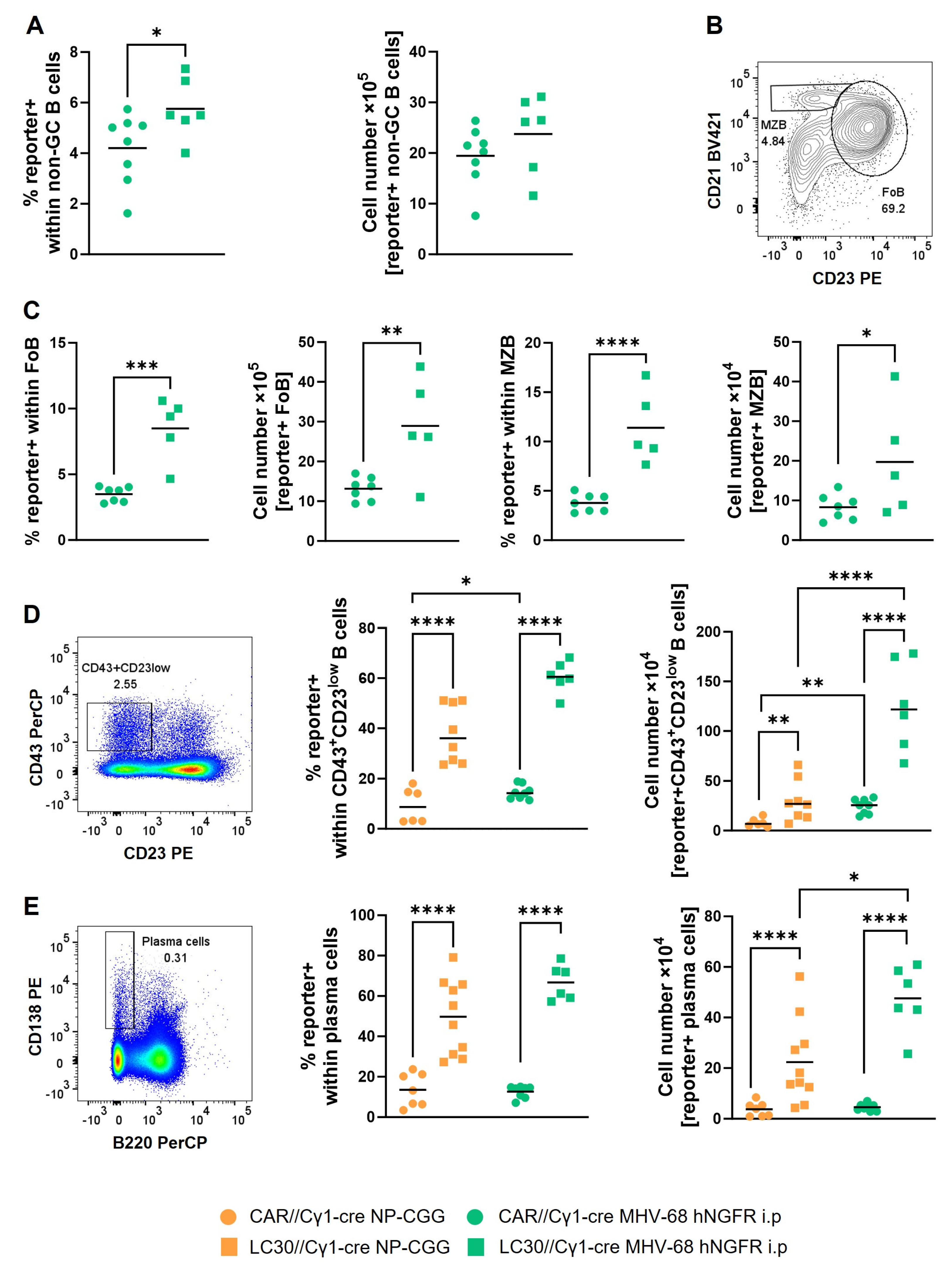
3.2. MHV-68 hNGFR Infection Results in Cre-Reporter Expression in GC B Cells and Isotype-Switched Cells
3.3. The Insertion of an EBV Gene into MHV-68 to Investigate Its Effect in the Context of a Natural Infection
3.4. The Induction of Constitutively Active CD30 Signaling via MHV-68 Infection Results in the Expansion of CD43+CD23low B Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Böni, M.; Rieble, L.; Münz, C. Co-Infection of the Epstein-Barr Virus and the Kaposi Sarcoma-Associated Herpesvirus. Viruses 2022, 14, 2709. [Google Scholar] [CrossRef]
- Weniger, M.A.; Küppers, R. Molecular biology of Hodgkin lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef]
- Ross, A.M.; Leahy, C.I.; Neylon, F.; Steigerova, J.; Flodr, P.; Navratilova, M.; Urbankova, H.; Vrzalikova, K.; Mundo, L.; Lazzi, S.; et al. Epstein-Barr Virus and the Pathogenesis of Diffuse Large B-Cell Lymphoma. Life 2023, 13, 521. [Google Scholar] [CrossRef]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Wang, Y.; Tibbetts, S.A.; Krug, L.T. Conquering the Host: Determinants of Pathogenesis Learned from Murine Gammaherpesvirus 68. Annu. Rev. Virol. 2021, 8, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 151–209. [Google Scholar] [PubMed]
- Damania, B.; Kenney, S.C.; Raab-Traub, N. Epstein-Barr virus: Biology and clinical disease. Cell 2022, 185, 3652–3670. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef]
- Rastelli, J.; Hömig-Hölzel, C.; Seagal, J.; Müller, W.; Hermann, A.C.; Rajewsky, K.; Zimber-Strobl, U. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 2008, 111, 1448–1455. [Google Scholar] [CrossRef]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). In Epstein Barr Virus Volume 2: One Herpes Virus: Many Diseases; Münz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 119–149. [Google Scholar]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 9150–9154. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). In Epstein Barr Virus Volume 2: One Herpes Virus: Many Diseases; Münz, C., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 151–180. [Google Scholar]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef]
- Sausen, D.G.; Basith, A.; Muqeemuddin, S. EBV and Lymphomagenesis. Cancers 2023, 15, 2133. [Google Scholar] [CrossRef]
- Willer, D.O.; Speck, S.H. Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 2003, 77, 8310–8321. [Google Scholar] [CrossRef]
- Zelazowska, M.A.; McBride, K.; Krug, L.T. Dangerous Liaisons: Gammaherpesvirus Subversion of the Immunoglobulin Repertoire. Viruses 2020, 12, 788. [Google Scholar] [CrossRef]
- Adler, H.; Messerle, M.; Wagner, M.; Koszinowski, U.H. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 2000, 74, 6964–6974. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Tiacci, E.; Schneider, S.; Arnolds, J.; Rüschenbaum, S.; Duppach, J.; Seifert, M.; Döring, C.; Hansmann, M.L.; Küppers, R. Human CD30+ B cells represent a unique subset related to Hodgkin lymphoma cells. J. Clin. Investig. 2018, 128, 2996–3007. [Google Scholar] [CrossRef] [PubMed]
- Sperling, S.; Fiedler, P.; Lechner, M.; Pollithy, A.; Ehrenberg, S.; Schiefer, A.-I.; Kenner, L.; Feuchtinger, A.; Kühn, R.; Swinerd, G.; et al. Chronic CD30 signaling in B cells results in lymphomagenesis by driving the expansion of plasmablasts and B1 cells. Blood 2019, 133, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Susanibar-Adaniya, S.; Barta, S.K. 2021 Update on Diffuse large B cell lymphoma: A review of current data and potential applications on risk stratification and management. Am. J. Hematol. 2021, 96, 617–629. [Google Scholar] [CrossRef]
- Casola, S.; Cattoretti, G.; Uyttersprot, N.; Koralov, S.B.; Seagal, J.; Hao, Z.; Waisman, A.; Egert, A.; Ghitza, D.; Rajewsky, K. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc. Natl. Acad. Sci. USA 2006, 103, 7396–7401. [Google Scholar] [CrossRef]
- Henning, A.L.; Sampson, J.N.B.; McFarlin, B.K. Measurement of Low-Abundance Intracellular mRNA Using Amplified FISH Staining and Image-Based Flow Cytometry. Curr. Protoc. Cytom. 2016, 76, 7.46.1–7.46.8. [Google Scholar] [CrossRef]
- Bowden, R.J.; Simas, J.P.; Davis, A.J.; Efstathiou, S. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol. 1997, 78 Pt 7, 1675–1687. [Google Scholar] [CrossRef]
- Fournier, B.; Boutboul, D.; Bruneau, J.; Miot, C.; Boulanger, C.; Malphettes, M.; Pellier, I.; Dunogué, B.; Terrier, B.; Suarez, F.; et al. Rapid identification and characterization of infected cells in blood during chronic active Epstein-Barr virus infection. J. Exp. Med. 2020, 217, e20192262. [Google Scholar] [CrossRef]
- Heger, K.; Kober, M.; Rieß, D.; Drees, C.; de Vries, I.; Bertossi, A.; Roers, A.; Sixt, M.; Schmidt-Supprian, M. A novel Cre recombinase reporter mouse strain facilitates selective and efficient infection of primary immune cells with adenoviral vectors. Eur. J. Immunol. 2015, 45, 1614–1620. [Google Scholar] [CrossRef]
- Flach, B.; Steer, B.; Thakur, N.N.; Haas, J.; Adler, H. The M10 locus of murine gammaherpesvirus 68 contributes to both the lytic and the latent phases of infection. J. Virol. 2009, 83, 8163–8172. [Google Scholar] [CrossRef]
- El-Gogo, S.; Flach, B.; Staib, C.; Sutter, G.; Adler, H. In vivo attenuation of recombinant murine gammaherpesvirus 68 (MHV-68) is due to the expression and immunogenicity but not to the insertion of foreign sequences. Virology 2008, 380, 322–327. [Google Scholar] [CrossRef]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Kurth, J.; Spieker, T.; Wustrow, J.; Strickler, G.J.; Hansmann, L.M.; Rajewsky, K.; Küppers, R. EBV-infected B cells in infectious mononucleosis: Viral strategies for spreading in the B cell compartment and establishing latency. Immunity 2000, 13, 485–495. [Google Scholar] [CrossRef]
- Zelazowska, M.A.; Dong, Q.; Plummer, J.B.; Zhong, Y.; Liu, B.; Krug, L.T.; McBride, K.M. Gammaherpesvirus-infected germinal center cells express a distinct immunoglobulin repertoire. Life Sci. Alliance 2020, 3, e201900526. [Google Scholar] [CrossRef] [PubMed]
- Milho, R.; Smith, C.M.; Marques, S.; Alenquer, M.; May, J.S.; Gillet, L.; Gaspar, M.; Efstathiou, S.; Simas, J.P.; Stevenson, P.G. In vivo imaging of murid herpesvirus-4 infection. J. Gen. Virol. 2009, 90, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; Sacher, T.; Adler, H.; Jordan, S.; Koszinowski, U.H.; Ruzsics, Z. Systemic and local infection routes govern different cellular dissemination pathways during gammaherpesvirus infection in vivo. J. Virol. 2013, 87, 4596–4608. [Google Scholar] [CrossRef]
- Nash, A.A.; Dutia, B.M.; Stewart, J.P.; Davison, A.J. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.M.; Speck, S.H. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS ONE 2012, 7, e33230. [Google Scholar] [CrossRef]
- Toellner, K.M.; Luther, S.A.; Sze, D.M.; Choy, R.K.; Taylor, D.R.; MacLennan, I.C.; Acha-Orbea, H. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J. Exp. Med. 1998, 187, 1193–1204. [Google Scholar] [CrossRef]
- Jack, R.S.; Imanishi-Kari, T.; Rajewsky, K. Idiotypic analysis of the response of C57BL/6 mice to the (4-hydroxy-3-nitrophenyl)acetyl group. Eur. J. Immunol. 1977, 7, 559–565. [Google Scholar] [CrossRef]
- Sarawar, S.R.; Cardin, R.D.; Brooks, J.W.; Mehrpooya, M.; Tripp, R.A.; Doherty, P.C. Cytokine production in the immune response to murine gammaherpesvirus 68. J. Virol. 1996, 70, 3264–3268. [Google Scholar] [CrossRef]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Mégret, J.; Storck, S.; Reynaud, C.A.; Weill, J.C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Shinnakasu, R.; Inoue, T.; Kometani, K.; Moriyama, S.; Adachi, Y.; Nakayama, M.; Takahashi, Y.; Fukuyama, H.; Okada, T.; Kurosaki, T. Regulated selection of germinal-center cells into the memory B cell compartment. Nat. Immunol. 2016, 17, 861–869. [Google Scholar] [CrossRef]
- Kurth, J.; Hansmann, M.L.; Rajewsky, K.; Küppers, R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc. Natl. Acad. Sci. USA 2003, 100, 4730–4735. [Google Scholar] [CrossRef]
- Portis, T.; Dyck, P.; Longnecker, R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 102, 4166–4178. [Google Scholar] [CrossRef]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef]
- Frederico, B.; Chao, B.; May, J.S.; Belz, G.T.; Stevenson, P.G. A murid gamma-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 2014, 15, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Collins, C.M.; Mendel, J.B.; Iwakoshi, N.N.; Speck, S.H. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 2009, 5, e1000677. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; May, J.S.; Connor, V.; Efstathiou, S. Vaccination against a hit-and-run viral cancer. J. Gen. Virol. 2010, 91 Pt 9, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: Expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2407–2421. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rambold, U.; Sperling, S.; Chew, Z.; Wang, Y.; Steer, B.; Zeller, K.; Strobl, L.J.; Zimber-Strobl, U.; Adler, H. A Mouse Model to Study the Pathogenesis of γ-herpesviral Infections in Germinal Center B Cells. Cells 2023, 12, 2780. https://doi.org/10.3390/cells12242780
Rambold U, Sperling S, Chew Z, Wang Y, Steer B, Zeller K, Strobl LJ, Zimber-Strobl U, Adler H. A Mouse Model to Study the Pathogenesis of γ-herpesviral Infections in Germinal Center B Cells. Cells. 2023; 12(24):2780. https://doi.org/10.3390/cells12242780
Chicago/Turabian StyleRambold, Ursula, Stefanie Sperling, Zakir Chew, Yan Wang, Beatrix Steer, Krisztina Zeller, Lothar J. Strobl, Ursula Zimber-Strobl, and Heiko Adler. 2023. "A Mouse Model to Study the Pathogenesis of γ-herpesviral Infections in Germinal Center B Cells" Cells 12, no. 24: 2780. https://doi.org/10.3390/cells12242780