
Inhibition of Abl Kinase by Imatinib Can Rescue the Compromised Barrier Function of 22q11.2DS Patient-iPSC-Derived Blood–Brain Barriers

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
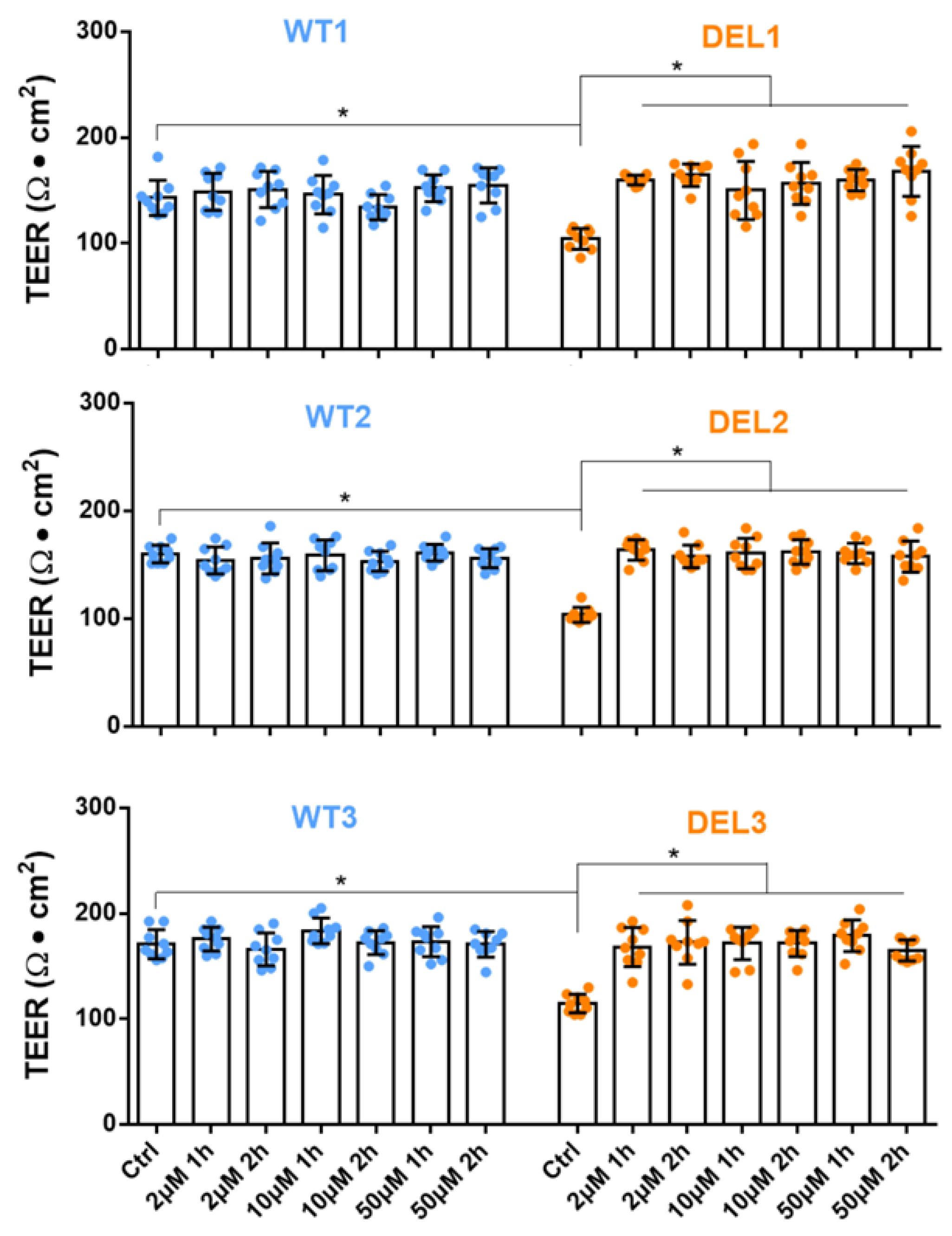
2.1. Generation of Induced BBB (iBBB) and Measurement of Its Trans-Endothelial Electrical Resistance (TEER)
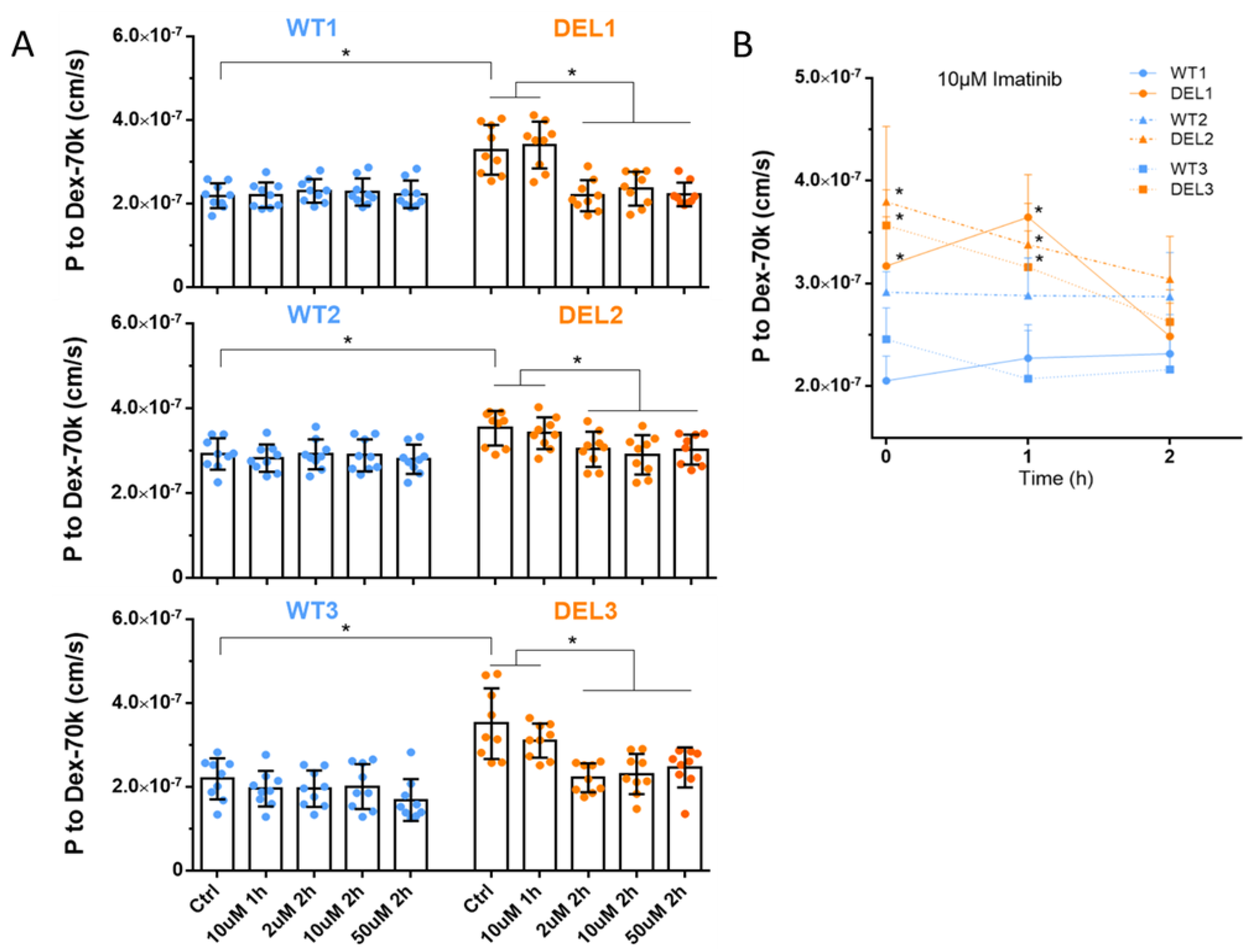
2.2. Determination of Solute Permeability (P) of iBBB
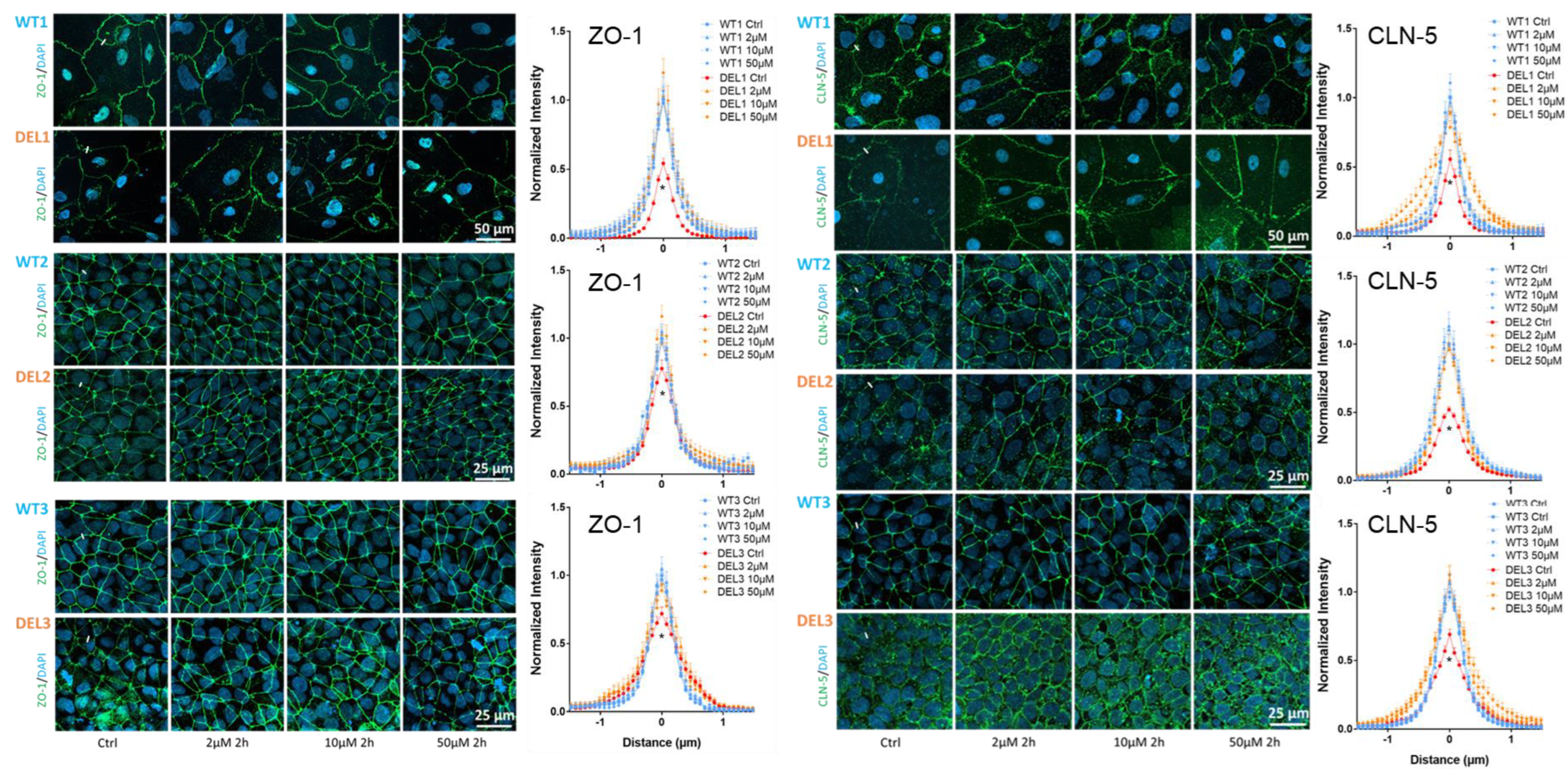
2.3. Immunostaining of Tight Junction Proteins and Glycocalyx of iBBB
2.4. Quantification of ZO-1, Claudin-5 (CLN-5) and HS (Glycocalyx)
2.5. Western Blotting
2.6. Total RNA Isolation and Bulk RNA Sequencing
2.7. Differential Expression Analysis
2.8. Gene Ontology Enrichment and KEGG Pathways Analysis
2.9. Treatment with Imatinib
2.10. Statistical Analysis
3. Results
3.1. Imatinib Restores the Barrier Function of 22q11.2DS iBBBs
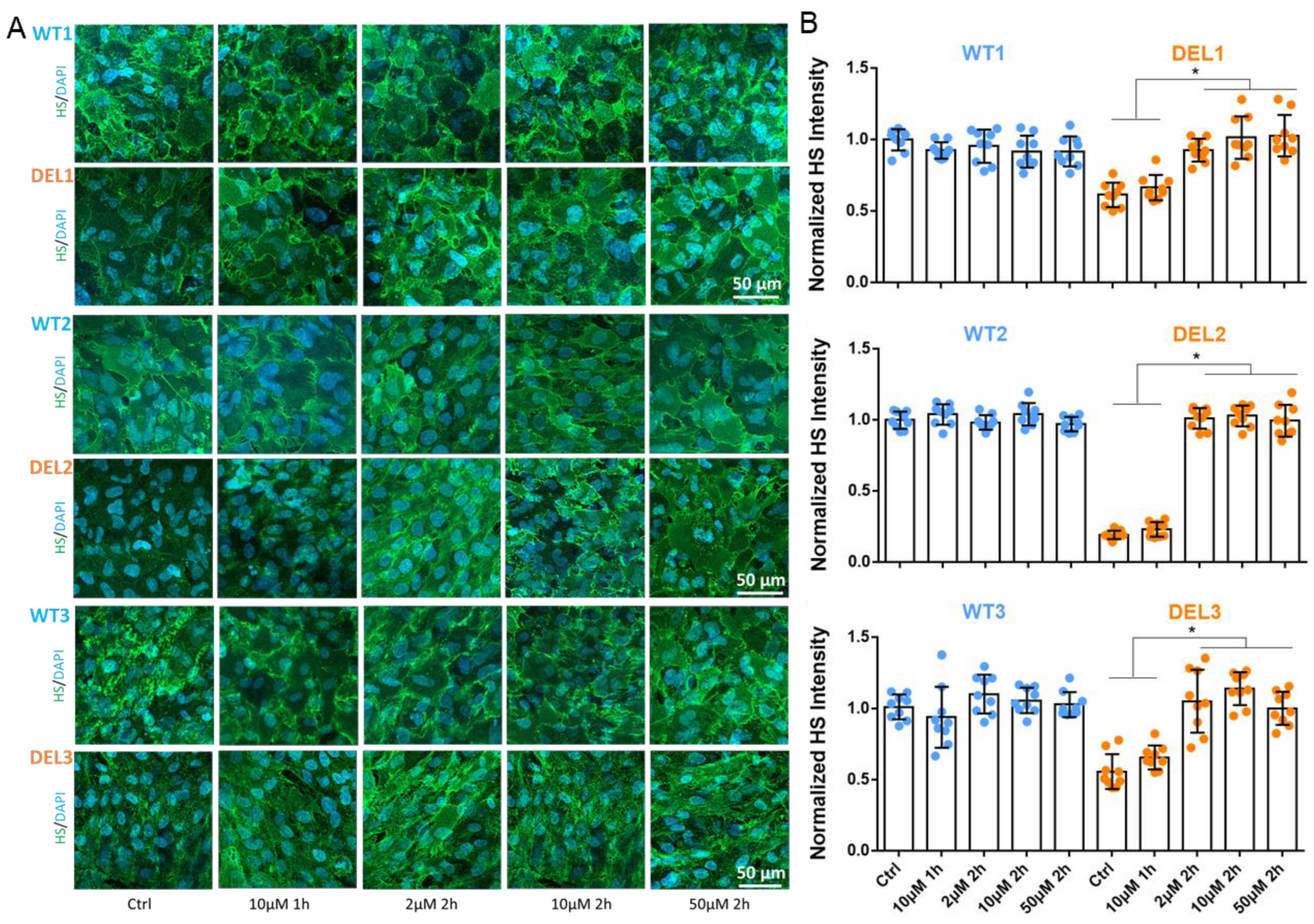
3.2. Imatinib Restores the Tight Junctions and Endothelial Glycocalyx of 22q11.2DS iBBBs
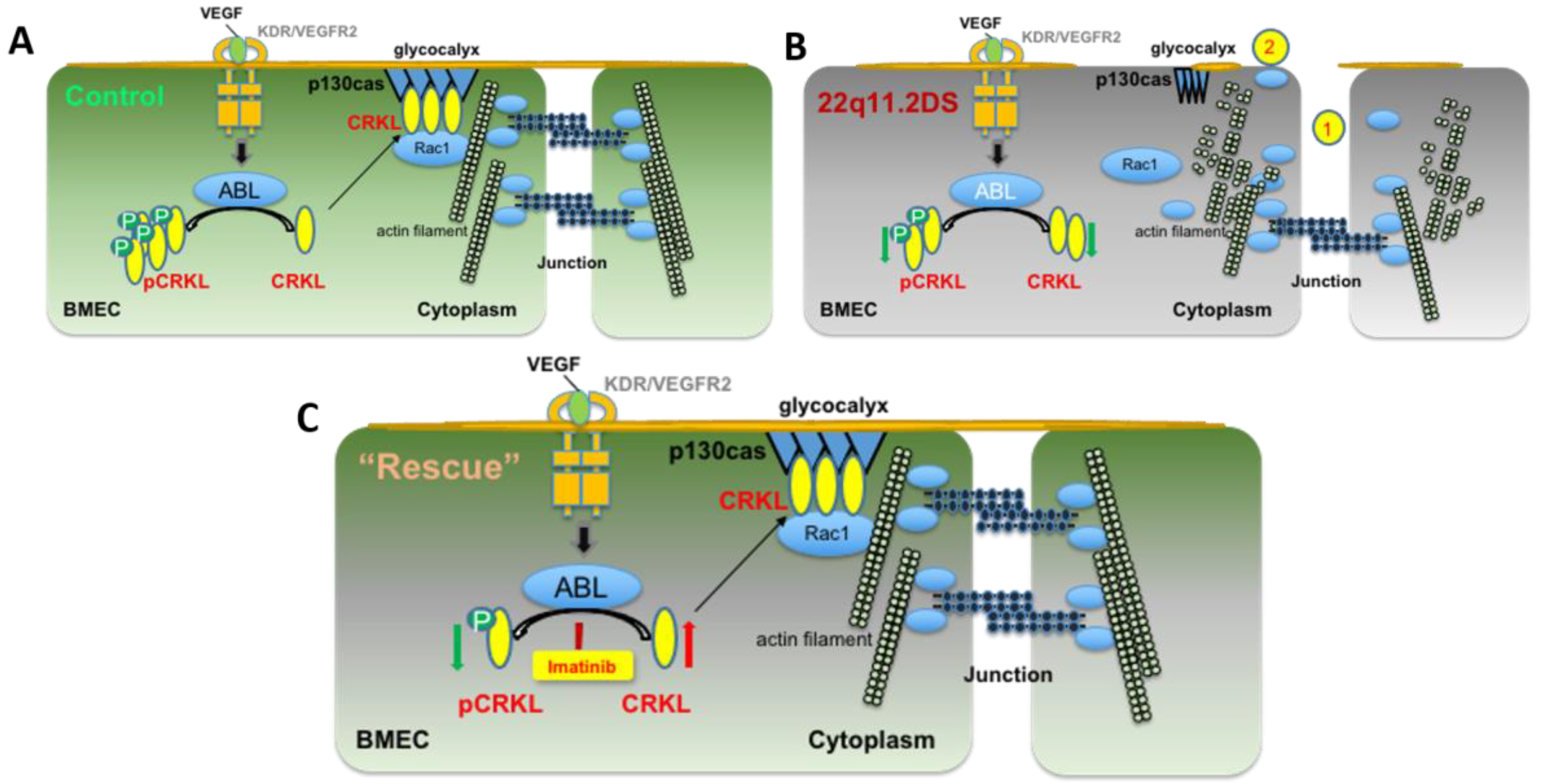
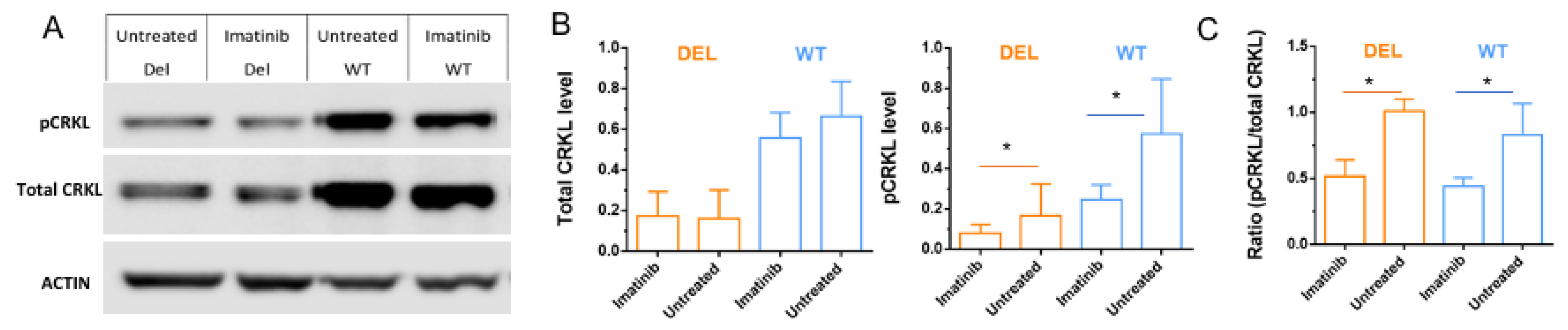
3.3. Imatinib Increases Active Forms of CRKL in HBMECs
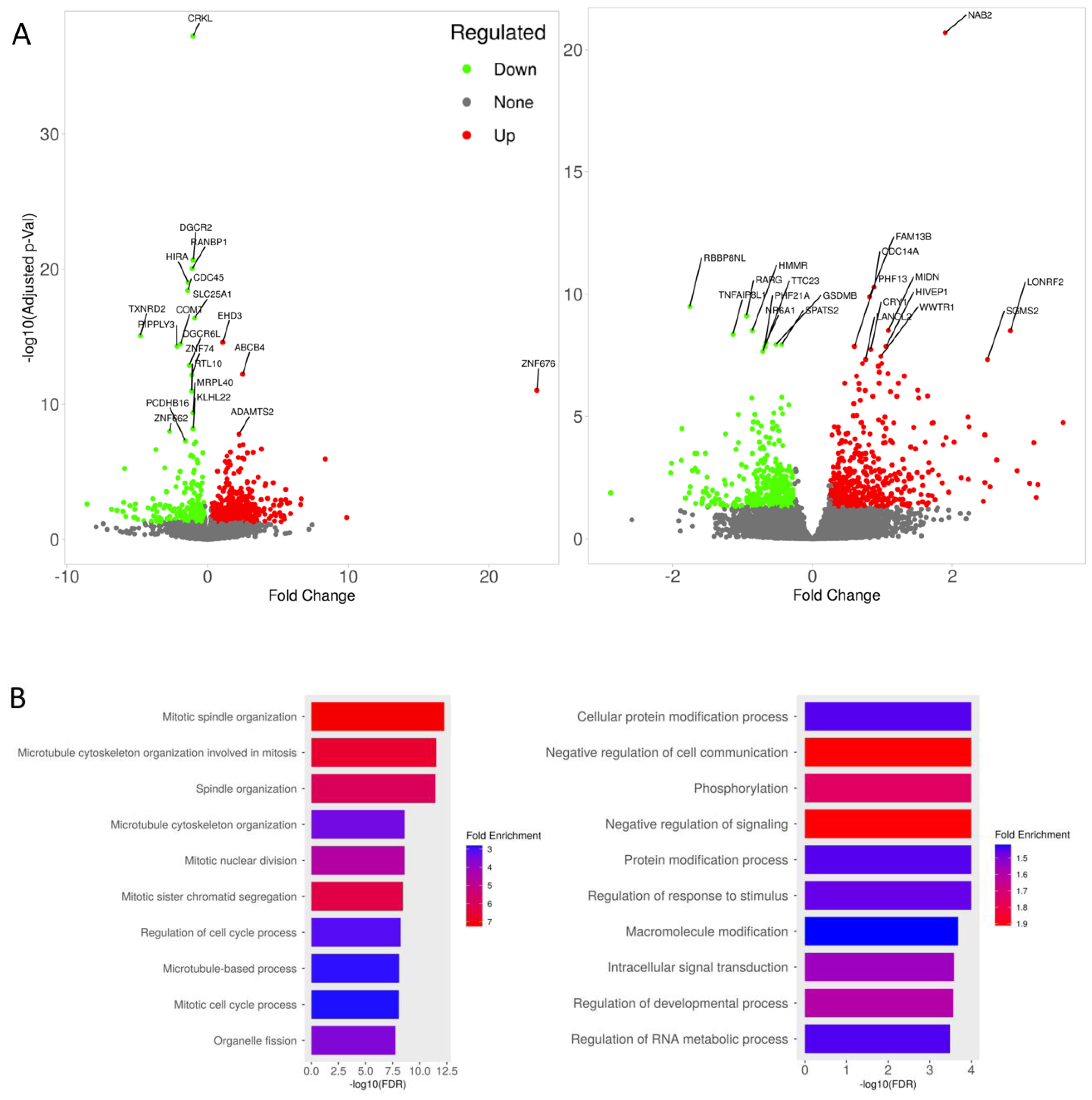
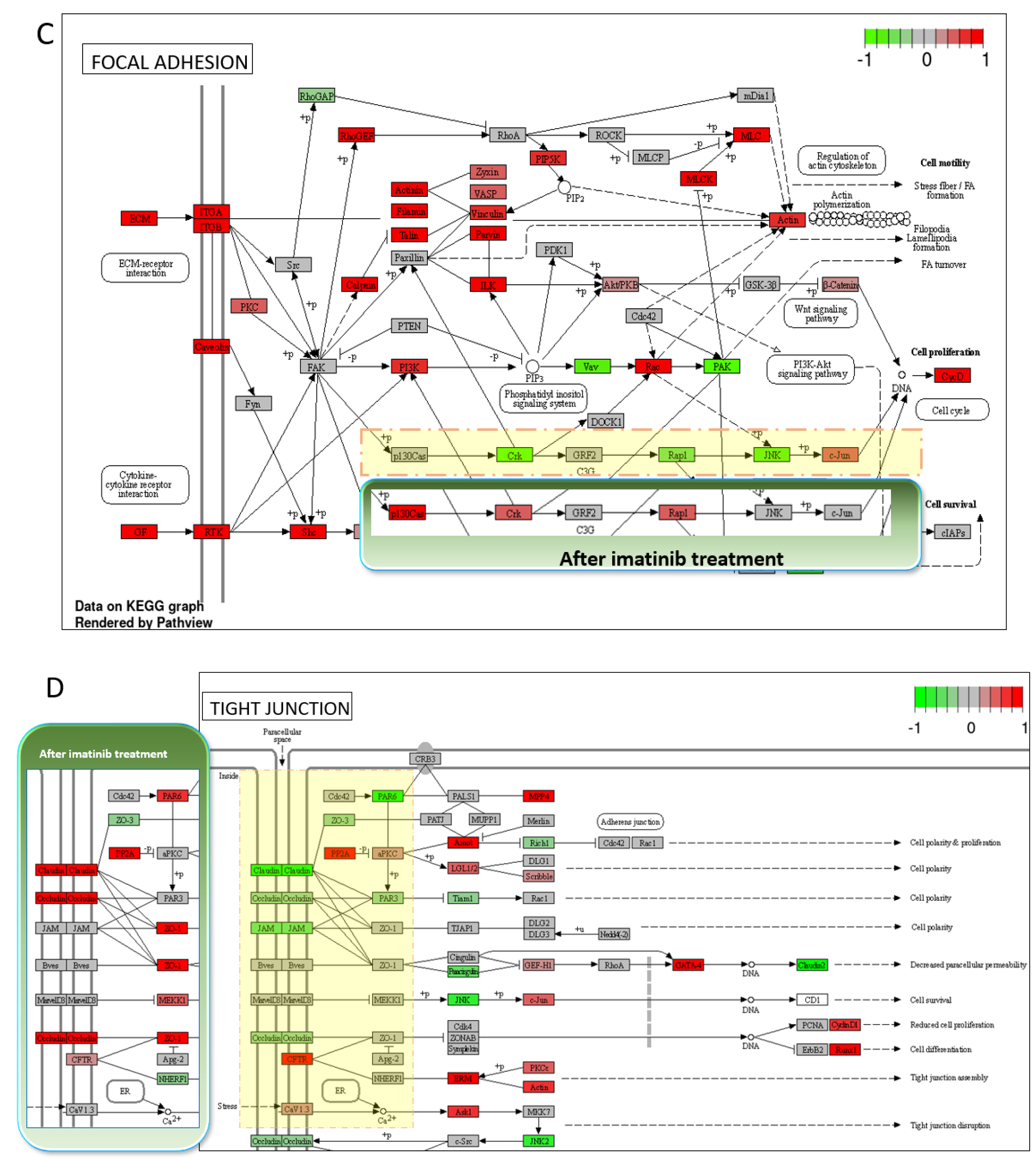
3.4. Transcriptomic Changes of iBBBs with Imatinib Treatment
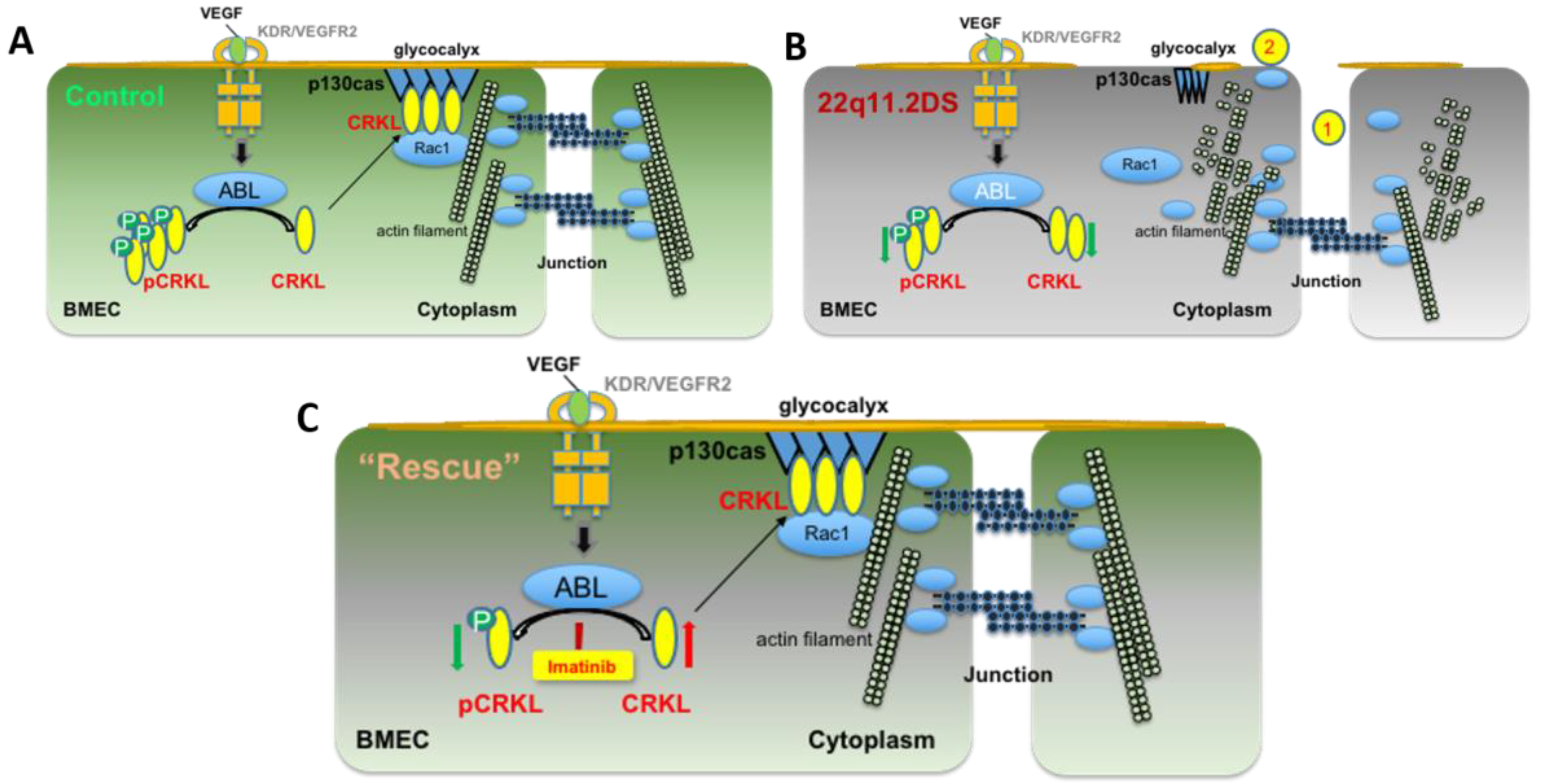
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benn, P.; Iyengar, S.; Crowley, T.B.; Zackai, E.H.; Burrows, E.K.; Moshkevich, S.; McDonald-McGinn, D.M.; Sullivan, K.E.; Demko, Z. Pediatric healthcare costs for patients with 22q11.2 deletion syndrome. Mol. Genet. Genomic. Med. 2017, 5, 631–638. [Google Scholar] [CrossRef]
- Henriksen, M.G.; Nordgaard, J.; Jansson, L.B. Genetics of Schizophrenia: Overview of Methods, Findings and Limitations. Front. Hum. Neurosci. 2017, 11, 322. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, A.; Lioncino, M.; Maratea, A.; Passariello, A.; Fusco, A.; Fratta, F.; Monda, E.; Caiazza, M.; Signore, G.; Esposito, A. Clinical manifestations of 22q11. 2 deletion syndrome. Heart Fail. Clin. 2022, 18, 155–164. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E. 22q11. 2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 1–19. [Google Scholar]
- Li, Y.; Xia, Y.; Zhu, H.; Luu, E.; Huang, G.; Sun, Y.; Sun, K.; Markx, S.; Leong, K.W.; Xu, B.; et al. Investigation of Neurodevelopmental Deficits of 22 q11.2 Deletion Syndrome with a Patient-iPSC-Derived Blood–Brain Barrier Model. Cells 2021, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Shalev, H.; Serlin, Y.; Friedman, A. Breaching the blood-brain barrier as a gate to psychiatric disorder. Cardiovasc. Psychiatry Neurol. 2009, 2009, 278531. [Google Scholar] [CrossRef] [Green Version]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uranova, N.A.; Zimina, I.S.; Vikhreva, O.V.; Krukov, N.O.; Rachmanova, V.I.; Orlovskaya, D.D. Ultrastructural damage of capillaries in the neocortex in schizophrenia. World J. Biol. Psychiatry 2010, 11, 567–578. [Google Scholar] [CrossRef]
- Uranova, N.; Zimina, I.; Vikhreva, O.; Rachmanova, V.; Klintsova, A.; Orlovskaya, D. Reduced capillary density in the prefrontal cortex in schizophrenia. Am. J. Med. Sci. Med. 2013, 1, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Vostrikov, V.; Orlovskaya, D.; Uranova, N. Deficit of pericapillary oligodendrocytes in the prefrontal cortex in schizophrenia. World J. Biol. Psychiatry 2008, 9, 34–42. [Google Scholar] [CrossRef]
- Azmitia, E.C.; Saccomano, Z.T.; Alzoobaee, M.F.; Boldrini, M.; Whitaker-Azmitia, P.M. Persistent Angiogenesis in the Autism Brain: An Immunocytochemical Study of Postmortem Cortex, Brainstem and Cerebellum. J. Autism. Dev. Disord. 2016, 46, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Enwright Iii, J.F.; Huo, Z.; Arion, D.; Corradi, J.P.; Tseng, G.; Lewis, D.A. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol. Psychiatry 2018, 23, 1606–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomelli, M.; Kumar, R.; Soresina, A.; Tamassia, N.; Lorenzini, T.; Moratto, D.; Gasperini, S.; Cassatella, M.; Plebani, A.; Lougaris, V.; et al. Reduction of CRKL expression in patients with partial DiGeorge syndrome is associated with impairment of T-cell functions. J. Allergy Clin. Immunol. 2016, 138, 229–240.e223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamorte, L.; Royal, I.; Naujokas, M.; Park, M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol. Biol. Cell 2002, 13, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.; Koptyra, M.; Curran, T. Fibroblast Growth Requires CT10 Regulator of Kinase (Crk) and Crk-like (CrkL)*. J. Biol. Chem. 2016, 291, 26273–26290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birge, R.B.; Kalodimos, C.; Inagaki, F.; Tanaka, S. Crk and CrkL adaptor proteins: Networks for physiological and pathological signaling. Cell Commun. Signal. 2009, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Chislock, E.M.; Pendergast, A.M. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS ONE 2013, 8, e85231. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.N.; Aman, J.; van Nieuw Amerongen, G.P.; Dudek, S.M. Targeting Abl kinases to regulate vascular leak during sepsis and acute respiratory distress syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Wood, L.J.; Agarwal, A.; O’Hare, T.; Elsea, C.R.; Griswold, I.J.; Deininger, M.W.; Imamoto, A.; Druker, B.J. A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors. Cancer Res. 2010, 70, 7325–7335. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [Green Version]
- Waller, C.F. Imatinib mesylate. Recent Results Cancer Res. 2010, 184, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Rhee, C.K.; Yeo, C.D.; Kang, H.H.; Lee, D.G.; Lee, S.H.; Kim, J.W. Effect of tyrosine kinase inhibitors, imatinib and nilotinib, in murine lipopolysaccharide-induced acute lung injury during neutropenia recovery. Crit. Care 2013, 17, R114. [Google Scholar] [CrossRef] [Green Version]
- Aman, J.; van Bezu, J.; Damanafshan, A.; Huveneers, S.; Eringa, E.C.; Vogel, S.M.; Groeneveld, A.B.J.; Noordegraaf, A.V.; Hinsbergh, V.W.M.v.; Amerongen, G.P.v.N. Effective Treatment of Edema and Endothelial Barrier Dysfunction With Imatinib. Circulation 2012, 126, 2728–2738. [Google Scholar] [CrossRef] [Green Version]
- Letsiou, E.; Rizzo, A.N.; Sammani, S.; Naureckas, P.; Jacobson, J.R.; Garcia, J.G.; Dudek, S.M. Differential and opposing effects of imatinib on LPS- and ventilator-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L259–L269. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, P. Imatinib buys time for brain after stroke. Nat. Med. 2008, 14, 712–713. [Google Scholar] [CrossRef]
- Su, E.J.; Fredriksson, L.; Geyer, M.; Folestad, E.; Cale, J.; Andrae, J.; Gao, Y.; Pietras, K.; Mann, K.; Yepes, M.; et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 2008, 14, 731–737. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Mechanisms of Brain Edema Formation in Mouse Models of Intracerebral Hemorrhage; Loma Linda University Libraries: Loma Linda, CA, USA, 2011. [Google Scholar]
- Lin, M.; Pedrosa, E.; Hrabovsky, A.; Chen, J.; Puliafito, B.R.; Gilbert, S.R.; Zheng, D.; Lachman, H.M. Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11. 2 deletion. BMC Syst. Biol. 2016, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Stebbins, M.J.; Wilson, H.K.; Canfield, S.G.; Qian, T.; Palecek, S.P.; Shusta, E.V. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods 2016, 101, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Fu, B.M. Quantification of Malignant Breast Cancer Cell MDA-MB-231 Transmigration Across Brain and Lung Microvascular Endothelium. Ann. Biomed. Eng. 2016, 44, 2189–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.; Li, G.; Gil, E.S.; Lowe, T.L.; Fu, B.M. Effect of surface charge of immortalized mouse cerebral endothelial cell monolayer on transport of charged solutes. Ann. Biomed. Eng. 2010, 38, 1463–1472. [Google Scholar] [CrossRef]
- Fu, B.M. Quantification of In Vitro Blood-Brain Barrier Permeability. Methods Mol. Biol. 2022, 2375, 217–228. [Google Scholar] [CrossRef]
- Qian, T.; Maguire, S.E.; Canfield, S.G.; Bao, X.; Olson, W.R.; Shusta, E.V.; Palecek, S.P. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci. Adv. 2017, 3, e1701679. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Sun, Y.; Xia, Y.; Tarbell, J.M.; Fu, B.M. Endothelial surface glycocalyx (ESG) components and ultra-structure revealed by stochastic optical reconstruction microscopy (STORM). Biorheology 2019, 56, 77–88. [Google Scholar] [CrossRef]
- Xia, Y.; Li, Y.; Fu, B.M. Differential effects of vascular endothelial growth factor on glycocalyx of endothelial and tumor cells and potential targets for tumor metastasis. APL Bioeng. 2022, 6, 016101. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.M.; Tarbell, J.M. Mechano-sensing and transduction by endothelial surface glycocalyx: Composition, structure, and function. Wires Mech. Dis. 2013, 5, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.M.; Yang, J.; Cai, B.; Fan, J.; Zhang, L.; Zeng, M. Reinforcing endothelial junctions prevents microvessel permeability increase and tumor cell adhesion in microvessels in vivo. Sci. Rep. 2015, 5, 15697. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X. iDEP Web Application for RNA-Seq Data Analysis. Methods Mol. Biol. 2021, 2284, 417–443. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G., Jr.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef] [Green Version]
- Aman, J.; Peters, M.J.L.; Weenink, C.; Amerongen, G.P.v.N.; Noordegraaf, A.V. Reversal of Vascular Leak with Imatinib. Am. J. Respir. Crit. Care Med. 2013, 188, 1171–1173. [Google Scholar] [CrossRef]
- Crockett, A.M.; Ryan, S.K.; Vásquez, A.H.; Canning, C.; Kanyuch, N.; Kebir, H.; Ceja, G.; Gesualdi, J.; Zackai, E.; McDonald-McGinn, D.; et al. Disruption of the blood-brain barrier in 22q11.2 deletion syndrome. Brain 2021, 144, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Casas, B.S.; Vitória, G.; Prieto, C.P.; Casas, M.; Chacón, C.; Uhrig, M.; Ezquer, F.; Ezquer, M.; Rehen, S.K.; Palma, V. Schizophrenia-derived hiPSC brain microvascular endothelial-like cells show impairments in angiogenesis and blood-brain barrier function. Mol. Psychiatry 2022, 27, 3708–3718. [Google Scholar] [CrossRef] [PubMed]
- Taler, M.; Mekori-Domachevsky, E.; Vergaelen, E.; Claes, S.; Serur, Y.; Dar, S.; Levy-Shraga, Y.; Weizman, A.; Swillen, A.; Gothelf, D. Blood brain barrier permeability increases with age in individuals with 22q11.2 deletion syndrome. World J. Biol. Psychiatry 2022, 23, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.M. Transport Across the Blood–Brain Barrier. In Molecular, Cellular, and Tissue Engineering of the Vascular System; Springer Publishing: New York, NY, USA, 2018; pp. 235–259. [Google Scholar]
- Li, G.; Yuan, W.; Fu, B.M. A model for the blood–brain barrier permeability to water and small solutes. J. Biomech. 2010, 43, 2133–2140. [Google Scholar] [CrossRef]
- Li, G.; Fu, B.M. An Electrodiffusion Model for the Blood-Brain Barrier Permeability to Charged Molecules. J. Biomech. Eng. 2011, 133, 021002. [Google Scholar] [CrossRef] [PubMed]
- Su, E.; Fredriksson, L.; Kanzawa, M.; Moore, S.; Folestad, E.; Stevenson, T.; Nilsson, I.; Sashindranath, M.; Schielke, G.; Warnock, M.; et al. Imatinib treatment reduces brain injury in a murine model of traumatic brain injury. Front. Cell Neurosci. 2015, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Krafft, P.R.; Lekic, T.; Ma, Q.; Souvenir, R.; Zhang, J.H.; Tang, J. Imatinib preserves blood–brain barrier integrity following experimental subarachnoid hemorrhage in rats. J. Neurosci. Res. 2015, 93, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Katt, M.E.; Xu, Z.S.; Gerecht, S.; Searson, P.C. Human Brain Microvascular Endothelial Cells Derived from the BC1 iPS Cell Line Exhibit a Blood-Brain Barrier Phenotype. PLoS ONE 2016, 11, e0152105. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direction | GAGE Analysis: IMATINIB vs. CONTROL | Statistic | Genes | Adj. p-Value |
|---|---|---|---|---|
| Up | Focal adhesion | 4.28 | 168 | 3.8 × 10−3 |
| PI3K-Akt signaling pathway | 3.8134 | 257 | 1.2 × 10−2 | |
| Human cytomegalovirus infection | 3.5472 | 153 | 2.3 × 10−2 | |
| Tight junction | 3.4354 | 128 | 2.4 × 10−2 | |
| Human papillomavirus infection | 3.3749 | 259 | 2.4 × 10−2 | |
| Pathways in cancer | 3.3199 | 408 | 2.4 × 10−2 | |
| Salmonella infection | 3.246 | 208 | 2.6 × 10−2 | |
| Prostate cancer | 3.2287 | 82 | 2.6 × 10−2 | |
| Cytokine-cytokine receptor interaction | 3.2113 | 103 | 2.6 × 10−2 | |
| Kaposi sarcoma-associated herpesvirus infection | 3.1396 | 133 | 2.6 × 10−2 | |
| Amyotrophic lateral sclerosis | 3.1259 | 299 | 2.6 × 10−2 | |
| Regulation of actin cytoskeleton | 3.1104 | 166 | 2.6 × 10−2 | |
| Hepatitis C | 3.0765 | 114 | 2.8 × 10−2 | |
| Alzheimer disease | 2.9958 | 311 | 3.2 × 10−2 | |
| JAK-STAT signaling pathway | 2.9416 | 84 | 3.6 × 10−2 | |
| Leukocyte transendothelial migration | 2.9201 | 83 | 3.6 × 10−2 | |
| Ras signaling pathway | 2.8883 | 166 | 3.6 × 10−2 | |
| Prion disease | 2.8792 | 213 | 3.6 × 10−2 | |
| Small cell lung cancer | 2.7779 | 84 | 4.7 × 10−2 | |
| Proteoglycans in cancer | 2.7667 | 170 | 4.7 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Sun, Z.; Zhu, H.; Sun, Y.; Shteyman, D.B.; Markx, S.; Leong, K.W.; Xu, B.; Fu, B.M. Inhibition of Abl Kinase by Imatinib Can Rescue the Compromised Barrier Function of 22q11.2DS Patient-iPSC-Derived Blood–Brain Barriers. Cells 2023, 12, 422. https://doi.org/10.3390/cells12030422
Li Y, Sun Z, Zhu H, Sun Y, Shteyman DB, Markx S, Leong KW, Xu B, Fu BM. Inhibition of Abl Kinase by Imatinib Can Rescue the Compromised Barrier Function of 22q11.2DS Patient-iPSC-Derived Blood–Brain Barriers. Cells. 2023; 12(3):422. https://doi.org/10.3390/cells12030422
Chicago/Turabian StyleLi, Yunfei, Zhixiong Sun, Huixiang Zhu, Yan Sun, David B. Shteyman, Sander Markx, Kam W. Leong, Bin Xu, and Bingmei M. Fu. 2023. "Inhibition of Abl Kinase by Imatinib Can Rescue the Compromised Barrier Function of 22q11.2DS Patient-iPSC-Derived Blood–Brain Barriers" Cells 12, no. 3: 422. https://doi.org/10.3390/cells12030422
APA StyleLi, Y., Sun, Z., Zhu, H., Sun, Y., Shteyman, D. B., Markx, S., Leong, K. W., Xu, B., & Fu, B. M. (2023). Inhibition of Abl Kinase by Imatinib Can Rescue the Compromised Barrier Function of 22q11.2DS Patient-iPSC-Derived Blood–Brain Barriers. Cells, 12(3), 422. https://doi.org/10.3390/cells12030422