
T Cell Energy Metabolism Is a Target of Glucocorticoids in Mice, Healthy Humans, and MS Patients
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experimentation
2.2. Isolation and Culture of Murine T Cells
2.3. Isolation and Culture of Human T Cells
2.4. Quantitative RT-PCR
2.5. Flow Cytometry
2.6. Lactate Assay
2.7. T Cell Isolation from MS Patients
2.8. Statistics
3. Results
3.1. Dex Represses Metabolic Gene Expression in Murine T Cells in a GR-Dependent Manner
3.2. Control of Metabolic Gene Expression by Dex Is Unrelated to Their Pro-Apoptotic Activity
3.3. Dex Interferes with the Activation of T Cells from Healthy Human Subjects
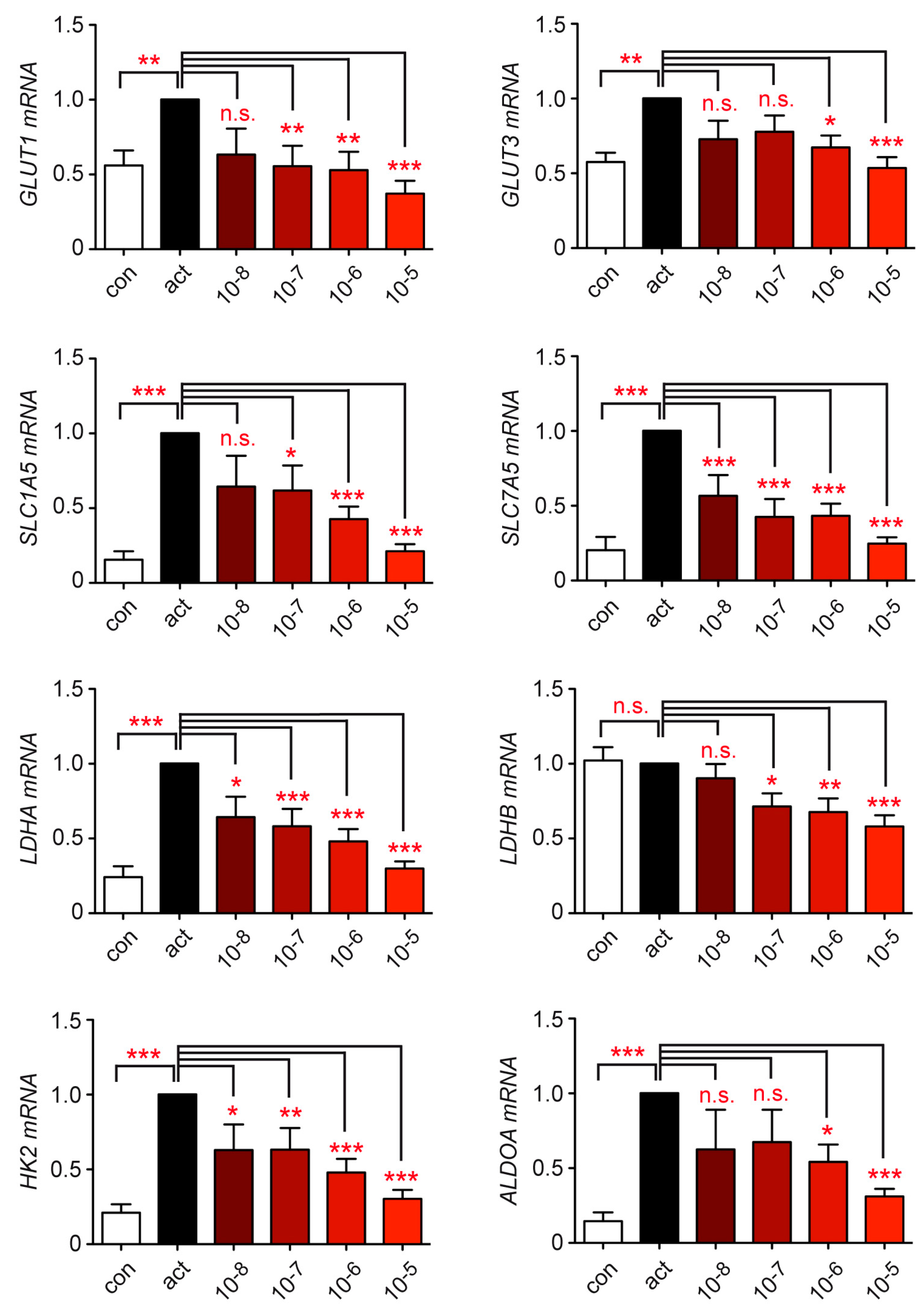
3.4. Metabolite Transport and Glycolysis in Human T Cells Are Repressed by Dex
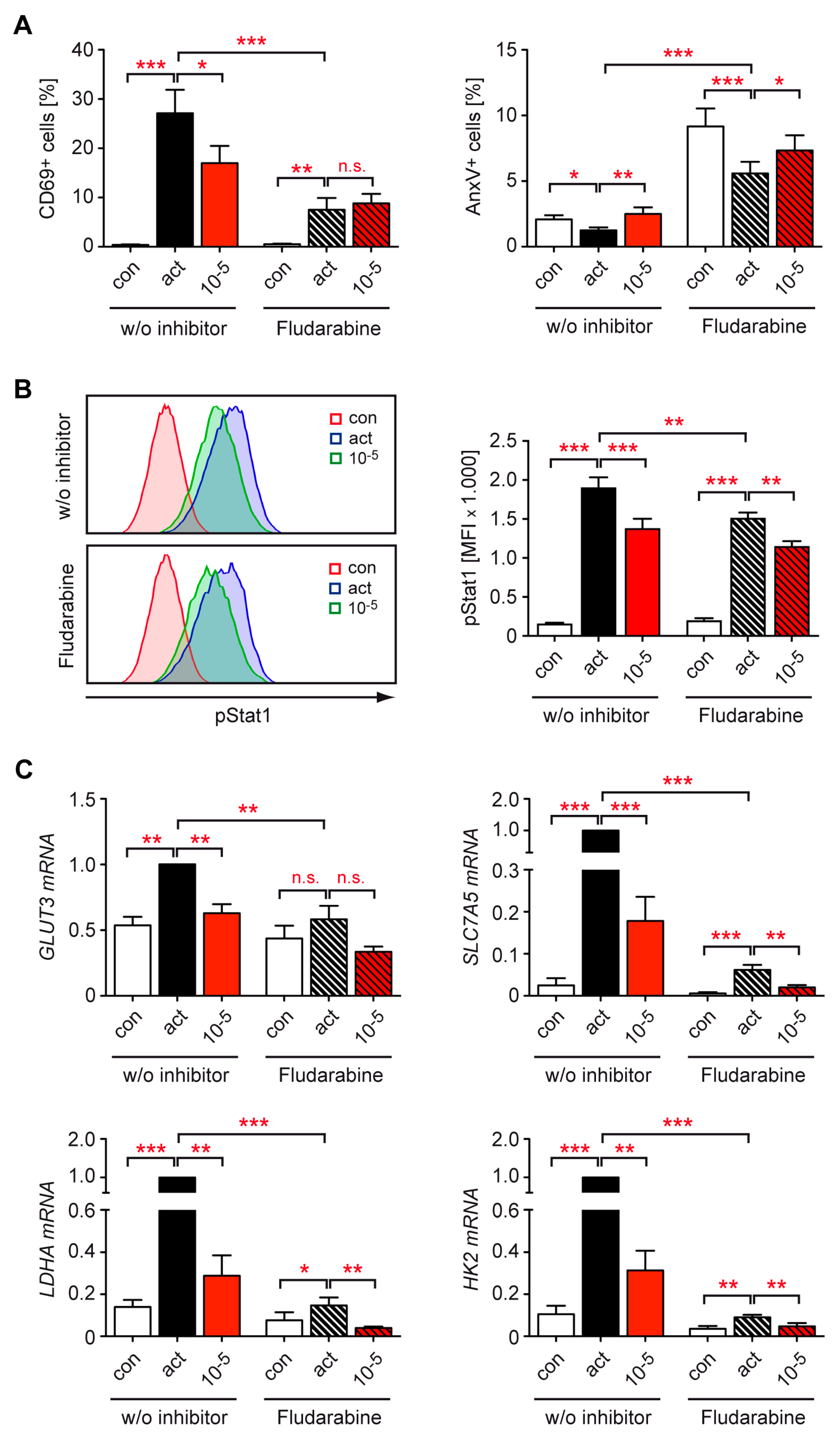
3.5. Stat1 Is Dispensable for the Inhibitory Effect of GCs on Metabolic Gene Expression in T Cells
3.6. Dex Represses T Cell Metabolism by Controlling mTOR Signaling
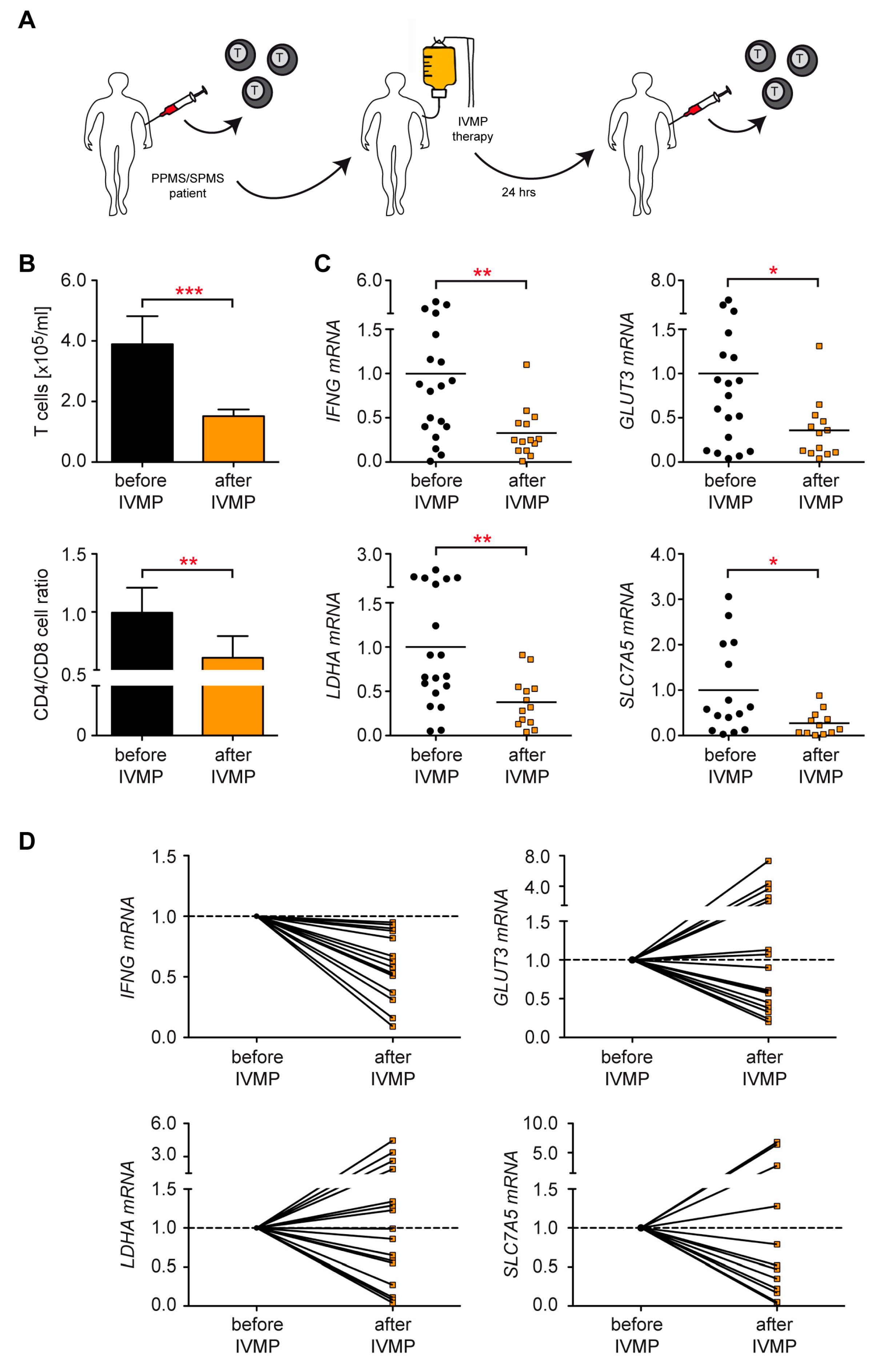
3.7. Control of T Cell Metabolism in MS Patients Receiving IVMP Therapy
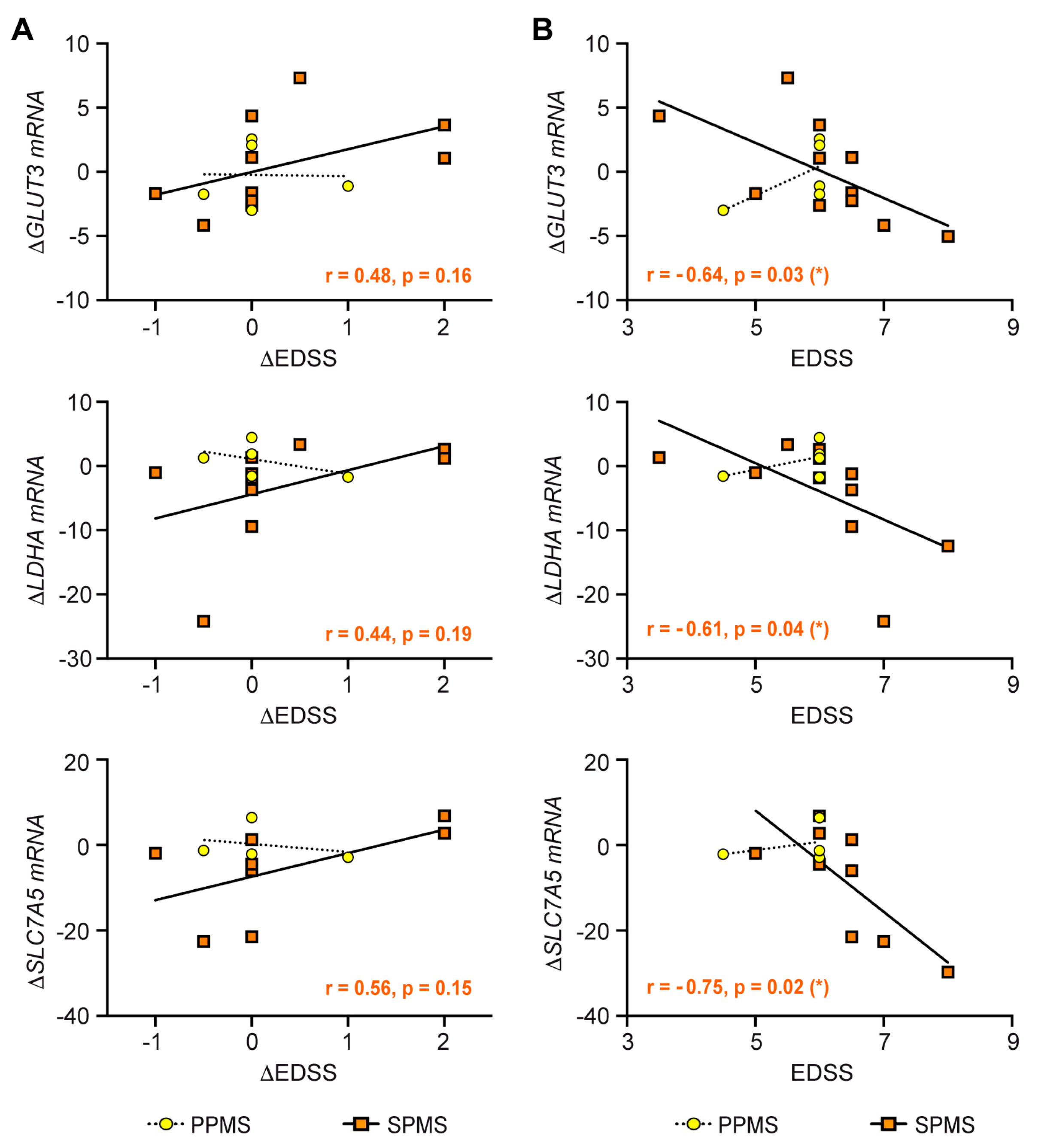
3.8. Correlation of Clinical Parameters with the Metabolic Control of T Cells by IVMP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef]
- Wüst, S.; van den Brandt, J.; Tischner, D.; Kleiman, A.; Tuckermann, J.P.; Gold, R.; Lühder, F.; Reichardt, H.M. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 8434–8443. [Google Scholar]
- Theiss-Suennemann, J.; Jorss, K.; Messmann, J.J.; Reichardt, S.D.; Montes-Cobos, E.; Lühder, F.; Tuckermann, J.P.; Wolff, H.A.; Dressel, R.; Gröne, H.J.; et al. Glucocorticoids attenuate acute graft-versus-host disease by suppressing the cytotoxic capacity of CD8(+) T cells. J. Pathol. 2015, 235, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Baschant, U.; Frappart, L.; Rauchhaus, U.; Bruns, L.; Reichardt, H.M.; Kamradt, T.; Brauer, R.; Tuckermann, J.P. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19317–19322. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Müller, N.; McPherson, K.G.; Reichardt, H.M. Glucocorticoids engage different signal transduction pathways to induce apoptosis in thymocytes and mature T cells. J. Immunol. 2006, 176, 1695–1702. [Google Scholar] [CrossRef] [Green Version]
- van den Brandt, J.; Wang, D.; Reichardt, H.M. Resistance of single-positive thymocytes to glucocorticoid-induced apoptosis is mediated by CD28 signaling. Mol. Endocrinol. 2004, 18, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischner, D.; Theiss, J.; Karabinskaya, A.; van den Brandt, J.; Reichardt, S.D.; Karow, U.; Herold, M.J.; Lühder, F.; Utermöhlen, O.; Reichardt, H.M. Acid sphingomyelinase is required for protection of effector memory T cells against glucocorticoid-induced cell death. J. Immunol. 2011, 187, 4509–4516. [Google Scholar] [CrossRef] [Green Version]
- Bier, J.; Steiger, S.M.; Reichardt, H.M.; Lühder, F. Protection of Antigen-Primed Effector T Cells From Glucocorticoid-Induced Apoptosis in Cell Culture and in a Mouse Model of Multiple Sclerosis. Front. Immunol. 2021, 12, 671258. [Google Scholar] [CrossRef]
- Wüst, S.; Tischner, D.; John, M.; Tuckermann, J.P.; Menzfeld, C.; Hanisch, U.K.; van den Brandt, J.; Lühder, F.; Reichardt, H.M. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS ONE 2009, 4, e8202. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, H.M.; Tuckermann, J.P.; Göttlicher, M.; Vujic, M.; Weih, F.; Angel, P.; Herrlich, P.; Schütz, G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. Embo. J. 2001, 20, 7168–7173. [Google Scholar] [CrossRef] [Green Version]
- Schweingruber, N.; Fischer, H.J.; Fischer, L.; van den Brandt, J.; Karabinskaya, A.; Labi, V.; Villunger, A.; Kretzschmar, B.; Huppke, P.; Simons, M.; et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014, 127, 713–729. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Fischer, H.J.; Tischner, D.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. J. Immunol. 2013, 190, 4360–4370. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Gold, R.; Schonrock, L.; Zettl, U.K.; Hartung, H.P.; Toyka, K.V. T-cell apoptosis in situ in experimental autoimmune encephalomyelitis following methylprednisolone pulse therapy. Brain 2000, 123, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Green, D.R. Metabolic checkpoints in activated T cells. Nat. Immunol. 2012, 13, 907–915. [Google Scholar] [CrossRef]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.; Opipari, A.W., Jr.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, C.K.; Andraski, A.B.; Spolski, R.; Li, P.; Kazemian, M.; Oh, J.; Samsel, L.; Swanson, P.A., 2nd; McGavern, D.B.; Sampaio, E.P.; et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9394–9399. [Google Scholar] [CrossRef] [Green Version]
- Stoycheva, D.; Deiser, K.; Starck, L.; Nishanth, G.; Schluter, D.; Uckert, W.; Schuler, T. IFN-gamma regulates CD8+ memory T cell differentiation and survival in response to weak, but not strong, TCR signals. J. Immunol. 2015, 194, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161 e145. [Google Scholar] [CrossRef]
- Bishop, E.L.; Gudgeon, N.; Dimeloe, S. Control of T Cell Metabolism by Cytokines and Hormones. Front. Immunol. 2021, 12, 653605. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, Q.T.; Lee, J.; Lee, S.H.; Janocha, A.; Kim, S.; Le, H.T.; Dvorina, N.; Weiss, K.; Cameron, M.J.; et al. Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity 2020, 53, 581–596 e585. [Google Scholar] [CrossRef]
- Hou, Y.; Xie, J.; Wang, S.; Li, D.; Wang, L.; Wang, H.; Ni, X.; Leng, S.; Li, G.; Hou, M.; et al. Glucocorticoid receptor modulates myeloid-derived suppressor cell function via mitochondrial metabolism in immune thrombocytopenia. Cell Mol. Immunol. 2022, 19, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, A.; Sugiyama, D.; Maeda, Y.; Warner, A.B.; Panageas, K.S.; Ito, S.; Togashi, Y.; Sakai, C.; Wolchok, J.D.; Nishikawa, H. Selective inhibition of low-affinity memory CD8(+) T cells by corticosteroids. J. Exp. Med. 2019, 216, 2701–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, A.; Suzuki, J.; Kuwahara, M.; Matsumoto, A.; Nomura, S.; Soga, T.; Yorozuya, T.; Yamashita, M. Glucocorticoid imprints a low glucose metabolism onto CD8 T cells and induces the persistent suppression of the immune response. Biochem. Biophys. Res. Commun. 2022, 588, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Haidinger, M.; Katholnig, K.; Kopecky, C.; Poglitsch, M.; Lassnig, C.; Rosner, M.; Zlabinger, G.J.; Hengstschlager, M.; Muller, M.; et al. Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells. Blood 2011, 117, 4273–4283. [Google Scholar] [CrossRef] [Green Version]
- Gayer, F.A.; Fichtner, A.; Legler, T.J.; Reichardt, H.M. A Coculture Model Mimicking the Tumor Microenvironment Unveils Mutual Interactions between Immune Cell Subtypes and the Human Seminoma Cell Line TCam-2. Cells 2022, 11, 885. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Lühder, F.; Wiegers, G.J.; Reichardt, H.M. A flow cytometric approach to study glucocorticoid receptor expression in immune cell subpopulations of genetically engineered mice. Immunol. Lett. 2021, 233, 68–79. [Google Scholar] [CrossRef]
- Fischer, H.J.; Sie, C.; Schumann, E.; Witte, A.K.; Dressel, R.; van den Brandt, J.; Reichardt, H.M. The Insulin Receptor Plays a Critical Role in T Cell Function and Adaptive Immunity. J. Immunol. 2017, 198, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.R.; Ashhurst, T.M.; West, P.K.; Viengkhou, B.; King, N.J.C.; Campbell, I.L.; Hofer, M.J. Contribution of STAT1 to innate and adaptive immunity during type I interferon-mediated lethal virus infection. PLoS Pathog. 2020, 16, e1008525. [Google Scholar] [CrossRef] [Green Version]
- Ballegeer, M.; Van Looveren, K.; Timmermans, S.; Eggermont, M.; Vandevyver, S.; Thery, F.; Dendoncker, K.; Souffriau, J.; Vandewalle, J.; Van Wyngene, L.; et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J. Clin. Investig. 2018, 128, 3265–3279. [Google Scholar] [CrossRef]
- Colombetti, S.; Basso, V.; Mueller, D.L.; Mondino, A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J. Immunol. 2006, 176, 2730–2738. [Google Scholar] [CrossRef] [Green Version]
- Milligan, N.M.; Newcombe, R.; Compston, D.A. A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: 1. Clinical effects. J. Neurol. Neurosurg Psychiatry 1987, 50, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozakbas, S.; Cinar, B.P.; Oz, D.; Kosehasanogullari, G.; Kursun, B.B.; Kahraman, T. Monthly Pulse Methylprednisolone Therapy is Effective in Preventing Permanent Disease Progression in Secondary Progressive Multiple Sclerosis. Noro. Psikiyatr. Ars. 2019, 56, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Ratzer, R.; Iversen, P.; Bornsen, L.; Dyrby, T.B.; Christensen, J.R.; Ammitzboll, C.; Madsen, C.G.; Garde, E.; Lyksborg, M.; Andersen, B.; et al. Monthly oral methylprednisolone pulse treatment in progressive multiple sclerosis. Mult. Scler. J. 2016, 22, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Gold, R.; Berger, T.; Derfuss, T.; Linker, R.; Maurer, M.; Stangel, M.; Aktas, O.; Baum, K.; Berghoff, M.; et al. [Multiple sclerosis treatment consensus group (MSTCG): Position paper on disease-modifying treatment of multiple sclerosis 2021 (white paper)]. Nervenarzt 2021, 92, 773–801. [Google Scholar] [CrossRef]
- Rommer, P.S.; Buckow, K.; Ellenberger, D.; Friede, T.; Pitschnau-Michel, D.; Fuge, J.; Stuve, O.; Zettl, U.K.; German Multiple Sclerosis (MS) Registry of the German National MS Society (DMSG). Patients characteristics influencing the longitudinal utilization of steroids in multiple sclerosis--an observational study. Eur. J. Clin. Investig. 2015, 45, 587–593. [Google Scholar] [CrossRef]
- Iwanowski, P.; Losy, J. Immunological differences between classical phenothypes of multiple sclerosis. J. Neurol. Sci. 2015, 349, 10–14. [Google Scholar] [CrossRef]
- Schweingruber, N.; Reichardt, S.D.; Lühder, F.; Reichardt, H.M. Mechanisms of glucocorticoids in the control of neuroinflammation. J. Neuroendocr. 2012, 24, 174–182. [Google Scholar] [CrossRef]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Förster, I.; Habenicht, A.J.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390. [Google Scholar] [CrossRef]
- Schweingruber, N.; Haine, A.; Tiede, K.; Karabinskaya, A.; van den Brandt, J.; Wüst, S.; Metselaar, J.M.; Gold, R.; Tuckermann, J.P.; Reichardt, H.M.; et al. Liposomal encapsulation of glucocorticoids alters their mode of action in the treatment of experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 4310–4318. [Google Scholar] [CrossRef] [Green Version]
- Frijters, R.; Fleuren, W.; Toonen, E.J.; Tuckermann, J.P.; Reichardt, H.M.; van der Maaden, H.; van Elsas, A.; van Lierop, M.J.; Dokter, W.; de Vlieg, J.; et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genom. 2010, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Smedlund, K.B.; Sanchez, E.R.; Hinds, T.D., Jr. FKBP51 and the molecular chaperoning of metabolism. Trends Endocrinol. Metab. 2021, 32, 862–874. [Google Scholar] [CrossRef]
- Brasanac, J.; Hetzer, S.; Asseyer, S.; Kuchling, J.; Bellmann-Strobl, J.; Ritter, K.; Gamradt, S.; Scheel, M.; Haynes, J.D.; Brandt, A.U.; et al. Central stress processing, T-cell responsivity to stress hormones and disease severity in multiple sclerosis. Brain Commun. 2022, 4, fcac086. [Google Scholar] [CrossRef]
- Hu, X.; Li, W.P.; Meng, C.; Ivashkiv, L.B. Inhibition of IFN-gamma signaling by glucocorticoids. J. Immunol. 2003, 170, 4833–4839. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; Rosenstiel, P.; Hampe, J.; Nikolaus, S.; Groessner, B.; Schottelius, A.; Kuhbacher, T.; Hamling, J.; Folsch, U.R.; Seegert, D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 2002, 51, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Zhao, Y.; Kay, T.W.; Muglia, L.J. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl. Acad. Sci. USA 2011, 108, 9554–9559. [Google Scholar] [CrossRef] [Green Version]
- Flammer, J.R.; Dobrovolna, J.; Kennedy, M.A.; Chinenov, Y.; Glass, C.K.; Ivashkiv, L.B.; Rogatsky, I. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol. Cell Biol. 2010, 30, 4564–4574. [Google Scholar] [CrossRef] [Green Version]
- Frisullo, G.; Nociti, V.; Iorio, R.; Katia Patanella, A.; Bianco, A.; Caggiula, M.; Sancricca, C.; Tonali, P.A.; Mirabella, M.; Batocchi, A.P. Glucocorticoid treatment reduces T-bet and pSTAT1 expression in mononuclear cells from relapsing remitting multiple sclerosis patients. Clin. Immunol. 2007, 124, 284–293. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xia, R.; Dai, C.; Yan, S.; Xie, T.; Liu, B.; Gan, L.; Zhuang, Z.; Huang, Q. Dexamethasone inhibits the proliferation of tumor cells. Cancer Manag. Res. 2019, 11, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Hoepner, R.; Bagnoud, M.; Pistor, M.; Salmen, A.; Briner, M.; Synn, H.; Schrewe, L.; Guse, K.; Ahmadi, F.; Demir, S.; et al. Vitamin D increases glucocorticoid efficacy via inhibition of mTORC1 in experimental models of multiple sclerosis. Acta Neuropathol. 2019, 138, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, S.M.; Sasidhar, M.V.; Lagishetty, V.; Spence, R.D.; Umeda, E.; Ziehn, M.O.; Krieger, T.; Schulz, K.H.; Heesen, C.; Hewison, M.; et al. Dynamic development of glucocorticoid resistance during autoimmune neuroinflammation. J. Clin. Endocrinol. Metab. 2012, 97, E1402–E1410. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.S.; de Castro, M. Generalized and tissue specific glucocorticoid resistance. Mol. Cell Endocrinol. 2021, 530, 111277. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer-Heemsoth, L.; Mitschke, K.; Bier, J.; Schütz, K.; Villunger, A.; Legler, T.J.; Weber, M.S.; Lühder, F.; Reichardt, H.M. T Cell Energy Metabolism Is a Target of Glucocorticoids in Mice, Healthy Humans, and MS Patients. Cells 2023, 12, 450. https://doi.org/10.3390/cells12030450
Meyer-Heemsoth L, Mitschke K, Bier J, Schütz K, Villunger A, Legler TJ, Weber MS, Lühder F, Reichardt HM. T Cell Energy Metabolism Is a Target of Glucocorticoids in Mice, Healthy Humans, and MS Patients. Cells. 2023; 12(3):450. https://doi.org/10.3390/cells12030450
Chicago/Turabian StyleMeyer-Heemsoth, Leonie, Katja Mitschke, Jasmina Bier, Konstantin Schütz, Andreas Villunger, Tobias J. Legler, Martin S. Weber, Fred Lühder, and Holger M. Reichardt. 2023. "T Cell Energy Metabolism Is a Target of Glucocorticoids in Mice, Healthy Humans, and MS Patients" Cells 12, no. 3: 450. https://doi.org/10.3390/cells12030450
APA StyleMeyer-Heemsoth, L., Mitschke, K., Bier, J., Schütz, K., Villunger, A., Legler, T. J., Weber, M. S., Lühder, F., & Reichardt, H. M. (2023). T Cell Energy Metabolism Is a Target of Glucocorticoids in Mice, Healthy Humans, and MS Patients. Cells, 12(3), 450. https://doi.org/10.3390/cells12030450