

The Pursuit of the “Inside” of the Amyloid Hypothesis—Is C99 a Promising Therapeutic Target for Alzheimer’s Disease?
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Generation of C99 and Aβ
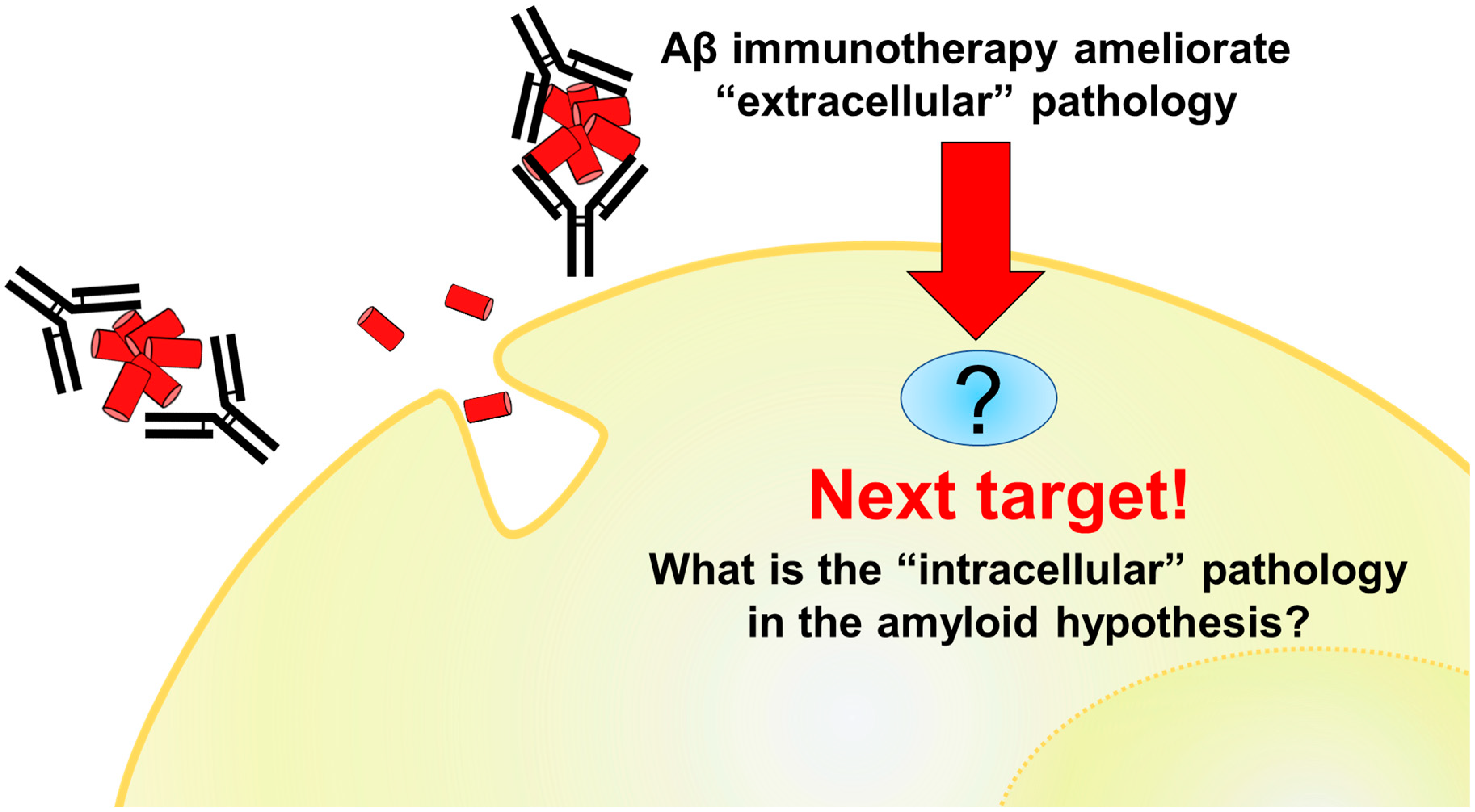
3. Potential and Limitations of Aβ Immunotherapy
4. C99 Accumulation in AD Pathology and Other Neurodegenerative Diseases
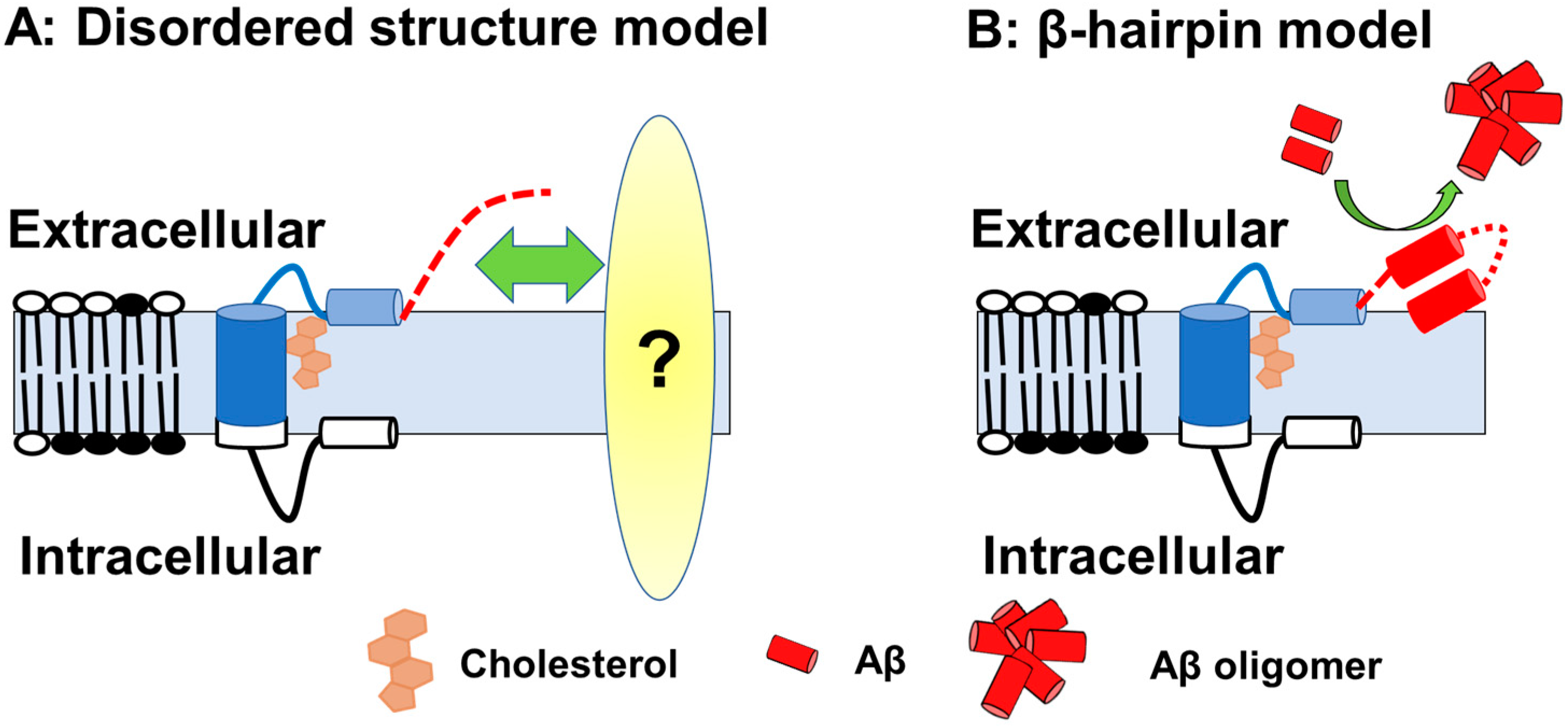
5. Structure and Metabolism of C99
6. Formation of Intracellular Pathology by C99 Accumulation
6.1. Endolysosomal Anomalies and C99
6.2. Autolysosomal Anomalies and C99
6.3. Intracellular Cholesterol Trafficking and C99
6.4. Effects of C99 Accumulation on Neuronal Function
7. Problems and Future Directions
7.1. BACE1 Targeting
7.2. C99 Targeting
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cummings, J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 2. [Google Scholar] [CrossRef]
- Cerejeira, J.; Lagarto, L.; Mukaetova-Ladinska, E.B. Behavioral and psychological symptoms of dementia. Front. Neurol. 2012, 3, 73. [Google Scholar] [CrossRef] [Green Version]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Afram, E.; Pardossi-Piquard, R.; Lauritzen, I. Is γ-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99? J. Biol. Chem. 2021, 296, 100489. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Makeen, H.A.; Albratty, M.; Abdellatif, A.A.H.; Dachani, S.R.; Bungau, S. Exploring the potential role of rab5 protein in endo-lysosomal impairment in Alzheimer’s disease. Biomed Pharm. 2022, 148, 112773. [Google Scholar] [CrossRef] [PubMed]
- Jakubauskienė, E.; Kanopka, A. Alternative Splicing and Hypoxia Puzzle in Alzheimer’s and Parkinson’s Diseases. Genes 2021, 12, 1272. [Google Scholar] [CrossRef]
- Peron, R.; Vatanabe, I.; Manzine, P.; Camins, A.; Cominetti, M. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the γ-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Lavoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, R.J.; Kellett, K.A.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis: A Focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Fukumoto, H.; Rosene, D.L.; Moss, M.B.; Raju, S.; Hyman, B.T.; Irizarry, M.C. β-Secretase Activity Increases with Aging in Human, Monkey, and Mouse Brain. Am. J. Pathol. 2004, 164, 719–725. [Google Scholar] [CrossRef]
- Fukumoto, H. β-Secretase Protein and Activity Are Increased in the Neocortex in Alzheimer Disease. Arch. Neurol. 2002, 59, 1381. [Google Scholar] [CrossRef]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Cheng, X.; He, P.; Lee, T.; Yao, H.; Li, R.; Shen, Y. High activities of BACE1 in brains with mild cognitive impairment. Am. J. Pathol. 2014, 184, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, N.; Takami, M.; Okochi, M.; Wada-Kakuda, S.; Fujiwara, H.; Tagami, S.; Funamoto, S.; Ihara, Y.; Morishima-Kawashima, M. γ-Secretase associated with lipid rafts: Multiple interactive pathways in the stepwise processing of β-carboxyl-terminal fragment. J. Biol. Chem. 2014, 289, 5109–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive Tripeptide and Tetrapeptide Release from the Transmembrane Domain of β-Carboxyl Terminal Fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [Green Version]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ 42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Mann, D.M.; Iwatsubo, T.; Ihara, Y.; Cairns, N.J.; Lantos, P.L.; Bogdanovic, N.; Lannfelt, L.; Winblad, B.; Maat-Schieman, M.L.; Rossor, M.N. Predominant deposition of amyloid-β 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am. J. Pathol. 1996, 148, 1257–1266. [Google Scholar]
- Hunter, S.; Brayne, C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol. Psychiatry 2018, 23, 81–93. [Google Scholar] [CrossRef]
- Tomita, T.; Maruyama, K.; Saido, T.C.; Kume, H.; Shinozaki, K.; Tokuhiro, S.; Capell, A.; Walter, J.; Grunberg, J.; Haass, C.; et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. USA 1997, 94, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e14. [Google Scholar] [CrossRef] [Green Version]
- De Jonghe, C. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001, 10, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, J.; Li, X.; Ma, L.; Hou, M.; Zhou, H.; Zhou, R. Based on molecular structures: Amyloid-β generation, clearance, toxicity and therapeutic strategies. Front. Mol. Neurosci. 2022, 15, 927530. [Google Scholar] [CrossRef]
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, J.; Zhang, Y.; Ubarretxena-Belandia, I.; Forth, S.; Lieberman, R.L.; Wang, C. Substrate-Enzyme Interactions in Intramembrane Proteolysis: Γ-Secretase as the Prototype. Front. Mol. Neurosci. 2020, 13, 65. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Taylor, H.A.; Przemylska, L.; Clavane, E.M.; Meakin, P.J. BACE1: More than just a β-secretase. Obes. Rev. 2022, 23, e13430. [Google Scholar] [CrossRef]
- Hu, X.; Hicks, C.W.; He, W.; Wong, P.; Macklin, W.B.; Trapp, B.D.; Yan, R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006, 9, 1520–1525. [Google Scholar] [CrossRef]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of peripheral nerve myelination by the β-secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef]
- Searfoss, G.H.; Jordan, W.H.; Calligaro, D.O.; Galbreath, E.J.; Schirtzinger, L.M.; Berridge, B.R.; Gao, H.; Higgins, M.A.; May, P.C.; Ryan, T.P. Adipsin, a Biomarker of Gastrointestinal Toxicity Mediated by a Functional γ-Secretase Inhibitor. J. Biol. Chem. 2003, 278, 46107–46116. [Google Scholar] [CrossRef] [Green Version]
- Savonenko, A.V.; Melnikova, T.; Laird, F.M.; Stewart, K.A.; Price, D.L.; Wong, P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5585–5590. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, D.; Zeller, M.; Cole, T.; Buttini, M.; McConlogue, L.; Sinha, S.; Freedman, S.; Morris, R.G.; Chen, K.S. BACE1 gene deletion: Impact on behavioral function in a model of Alzheimer’s disease. Neurobiol. Aging 2008, 29, 861–873. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A β antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2022, 388, 9–21. [Google Scholar] [CrossRef]
- Decourt, B.; Boumelhem, F.; Pope, E.D., 3rd; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jagust, W.J. Alzheimer Disease Spectrum: Syndrome and Etiology From Clinical and PET Imaging Perspectives. Neurology 2021, 96, 299–300. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Pera, M.; Alcolea, D.; Sanchez-Valle, R.; Guardia-Laguarta, C.; Colom-Cadena, M.; Badiola, N.; Suarez-Calvet, M.; Llado, A.; Barrera-Ocampo, A.A.; Sepulveda-Falla, D.; et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013, 125, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.O.Y.; Livesey, F.J. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018, 25, 3647–3660 e3642. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e255. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneshiro, N.; Komai, M.; Imaoka, R.; Ikeda, A.; Kamikubo, Y.; Saito, T.; Saido, T.C.; Tomita, T.; Hashimoto, T.; Iwatsubo, T.; et al. Lipid flippase dysfunction as a therapeutic target for endosomal anomalies in Alzheimer’s disease. iScience 2022, 25, 103869. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Pawlik, M.; Gandy, S.E.; Ehrlich, M.E.; Smiley, J.F.; Levy, E. Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol. Psychiatry 2017, 22, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Yabuki, C.; Seubert, P.; Schenk, D.; Hori, Y.; Ohtsuki, S.; Terasaki, T.; Hashimoto, T.; Iwatsubo, T. Aβ immunotherapy: Intracerebral sequestration of Aβ by an anti-Aβ monoclonal antibody 266 with high affinity to soluble Aβ. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11393–11398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watamura, N.; Sato, K.; Shiihashi, G.; Iwasaki, A.; Kamano, N.; Takahashi, M.; Sekiguchi, M.; Mihira, N.; Fujioka, R.; Nagata, K.; et al. An isogenic panel of App knock-in mouse models: Profiling β-secretase inhibition and endosomal abnormalities. Sci. Adv. 2022, 8, eabm6155. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.-W.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular Accumulation of Amyloidogenic Fragments of Amyloid-β Precursor Protein in Neurons with Niemann-Pick Type C Defects Is Associated with Endosomal Abnormalities. Am. J. Pathol. 2004, 164, 975–985. [Google Scholar] [CrossRef]
- Kosicek, M.; Malnar, M.; Goate, A.; Hecimovic, S. Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem. Biophys. Res. Commun. 2010, 393, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariati, I.; Masuelli, L.; Bei, R.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 6600. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Parker, C.C.; Pentchev, P.G.; Katz, D.; Ghetti, B.; D’Agostino, A.N.; Carstea, E.D. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995, 89, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann—Pick disease type C. Brain A J. Neurol. 1995, 118, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Chang, T.-Y.; Haass, C.; Ihara, Y. Accumulation and Aggregation of Amyloid β-Protein in Late Endosomes of Niemann-Pick Type C Cells. J. Biol. Chem. 2001, 276, 4454–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; Li, W.; Luo, Y.; Wang, D.; Zhu, C.; Huang, Z.X.; Tan, X. Exploring the differences between mouse mAβ(1-42) and human hAβ(1-42) for Alzheimer’s disease related properties and neuronal cytotoxicity. Chem. Commun. 2013, 49, 5865–5867. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.; Sharma, B.; Belfort, G. N-Terminal Hypothesis for Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 432–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) Familial Alzheimer Disease Mutations on Amyloid β-Protein Assembly and Toxicity. J. Biol. Chem. 2010, 285, 23186–23197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagnon de la Vega, M.; Giedraitis, V.; Michno, W.; Kilander, L.; Guner, G.; Zielinski, M.; Lowenmark, M.; Brundin, R.; Danfors, T.; Soderberg, L.; et al. The Uppsala APP deletion causes early onset autosomal dominant Alzheimer’s disease by altering APP processing and increasing amyloid β fibril formation. Sci. Transl. Med. 2021, 13, eabc6184. [Google Scholar] [CrossRef] [PubMed]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Pantelopulos, G.A.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Structure of APP-C991-99 and implications for role of extra-membrane domains in function and oligomerization. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1698–1708. [Google Scholar] [CrossRef]
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Xu, T.-H.; Melcher, K.; Xu, H.E. Defining the minimum substrate and charge recognition model of γ-secretase. Acta Pharmacol. Sin. 2017, 38, 1412–1424. [Google Scholar] [CrossRef] [Green Version]
- Struhl, G.; Adachi, A. Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol. Cell 2000, 6, 625–636. [Google Scholar] [CrossRef]
- Svedružić, Ž.M.; Jengić, V.Š.; Ostojić, L. Binding of different substrate molecules at the docking site and the active site of γ-secretase can trigger toxic events in sporadic and familial Alzheimer’s disease. bioRxiv 2022, 24, 1835. [Google Scholar] [CrossRef]
- Asai, M.; Yagishita, S.; Iwata, N.; Saido, T.C.; Ishiura, S.; Maruyama, K. An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model. FASEB J. 2011, 25, 3720–3730. [Google Scholar] [CrossRef]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.A.; Piper, S.; Baker, M.; Onstead, L.; Murphy, M.P.; Hardy, J.; Wang, R.; McGowan, E.; Golde, T.E. Expression of BRI-amyloid β peptide fusion proteins: A novel method for specific high-level expression of amyloid β peptides. Biochim. Biophys. Acta 2001, 1537, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcgowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Aβ42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Chakrabarty, P.; Hanna, A.; March, A.; Dickson, D.W.; Borchelt, D.R.; Golde, T.; Janus, C. Normal cognition in transgenic BRI2-Aβ mice. Mol. Neurodegener. 2013, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Suh, Y.-H. Neurotoxicity of a Carboxyl-Terminal Fragment of the Alzheimer’s Amyloid Precursor Protein. J. Neurochem. 2002, 67, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 1989, 245, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 2005, 26, 373–382. [Google Scholar] [CrossRef]
- Lewis, P.A. Vesicular dysfunction and pathways to neurodegeneration. Essays Biochem. 2021, 65, 941–948. [Google Scholar] [CrossRef]
- Kimura, N.; Yanagisawa, K. Traffic jam hypothesis: Relationship between endocytic dysfunction and Alzheimer’s disease. Neurochem. Int. 2018, 119, 35–41. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Cataldo, A.M.; Barnett, J.L.; Pieroni, C.; Nixon, R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: Neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 6142–6151. [Google Scholar] [CrossRef] [PubMed]
- Troncoso, J.C.; Cataldo, A.M.; Nixon, R.A.; Barnett, J.L.; Lee, M.K.; Checler, F.; Fowler, D.R.; Smialek, J.E.; Crain, B.; Martin, L.J.; et al. Neuropathology of preclinical and clinical lateonset Alzheimer’s disease. Ann. Neurol. 1998, 43, 673–676. [Google Scholar] [CrossRef]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein–mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Petanceska, S.; Peterhoff, C.M.; Terio, N.B.; Epstein, C.J.; Villar, A.; Carlson, E.J.; Staufenbiel, M.; Nixon, R.A. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6788–6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Rigoglioso, A.; Peterhoff, C.M.; Pawlik, M.; Sato, Y.; Bleiwas, C.; Stavrides, P.; Smiley, J.F.; Ginsberg, S.D.; Mathews, P.M.; et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: Role of APP-CTF. Neurobiol. Aging 2016, 39, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, N.; Araya, R.; Kamikubo, Y.; Kaneshiro, N.; Imaoka, R.; Jin, H.; Kashiyama, T.; Hashimoto, Y.; Kurosawa, M.; Uehara, T.; et al. TMEM30A is a candidate interacting partner for the β-carboxyl-terminal fragment of amyloid-β precursor protein in endosomes. PLoS ONE 2018, 13, e0200988. [Google Scholar] [CrossRef]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef]
- Xu, W.; Fang, F.; Ding, J.; Wu, C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic 2018, 19, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Song, C. The Emerging Role of Rab5 in Membrane Receptor Trafficking and Signaling Pathways. Biochem. Res. Int. 2020, 2020, 4186308. [Google Scholar] [CrossRef] [PubMed]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.-H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, G.; D’Ambrosio, J.M.; Dieudonné, T.; Čopič, A. Transport Pathways That Contribute to the Cellular Distribution of Phosphatidylserine. Front. Cell Dev. Biol. 2021, 9, 2412. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Jiang, Z.-J.; Gong, L.W. Phosphatidylserine is critical for vesicle fission during clathrin-mediated endocytosis. J. Neurochem. 2020, 152, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Hasegawa, J.; Chinnapen, D.; Inoue, T.; Okazaki, S.; Kato, R.; Wakatsuki, S.; Misaki, R.; Koike, M.; Uchiyama, Y.; et al. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc. Natl. Acad. Sci. USA 2011, 108, 15846–15851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Uchida, Y.; Emoto, K.; Umeda, M.; Kuge, O.; Taguchi, T.; Arai, H. Impaired retrograde membrane traffic through endosomes in a mutant CHO cell defective in phosphatidylserine synthesis. Genes Cells Devoted Mol. Cell. Mech. 2012, 17, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Uchida, Y.; Wang, J.; Matsudaira, T.; Nakagawa, T.; Kishimoto, T.; Mukai, K.; Inaba, T.; Kobayashi, T.; Molday, R.S.; et al. Transport through recycling endosomes requires EHD1 recruitment by a phosphatidylserine translocase. EMBO J. 2015, 34, 669–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsu, H.; Tanaka, G.; Segawa, K.; Suzuki, J.; Nagata, S.; Nakayama, K.; Shin, H.-W. Phospholipid Flippase Activities and Substrate Specificities of Human Type IV P-type ATPases Localized to the Plasma Membrane. J. Biol. Chem. 2014, 289, 33543–33556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Molday, R.S. Critical role of the β-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 2011, 286, 17205–17216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Kurata, S.; Nagata, S. The CDC50A extracellular domain is required for forming a functional complex with and chaperoning phospholipid flippases to the plasma membrane. J. Biol. Chem. 2018, 293, 2172–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Jiang, Y.; Zeng, S.; Yan, J.; Li, X.; Zhang, Y.; Zou, W.; Wang, X. Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS Genet. 2010, 6, e1001235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Im, E.; Jiang, Y.; Stavrides, P.; Darji, S.; Erdjument-Bromage, H.; Neubert, T.A.; Bordi, M.; Choi, J.Y.; Lee, J.-H.; Nixon, R.A. Lysosomal dysfunction in Down Syndrome and Alzheimer mouse models is caused by selective v-ATPase inhibition by Tyr682 phosphorylated APP βCTF. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Panahi, A.; Bandara, A.; Pantelopulos, G.A.; Dominguez, L.; Straub, J.E. Specific Binding of Cholesterol to C99 Domain of Amyloid Precursor Protein Depends Critically on Charge State of Protein. J. Phys. Chem. Lett. 2016, 7, 3535–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.R.; Montesinos, J.; Larrea, D.; Area-Gomez, E.; Pera, M. The silence of the fats: A MAM’s story about Alzheimer. Neurobiol. Dis. 2020, 145, 105062. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, N.; Fabiano, M.; Müller, U.C.; Walter, J. Carboxy-terminal fragment of amyloid precursor protein mediates lipid droplet accumulation upon γ-secretase inhibition. Biochem. Biophys. Res. Commun. 2021, 570, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease β-amyloid production. Biochim. Biophys. Acta 2010, 1801, 860–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, T.; Kaneko, K.; Okuno, M.; Wada, K.; Kashiyama, T.; Shimizu, H.; Akagi, T.; Hashikawa, T.; Nukina, N. Membrane microdomain switching: A regulatory mechanism of amyloid precursor protein processing. J. Cell Biol. 2008, 183, 339–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabelo, N.; Martin, V.; Marin, R.; Moreno, D.; Ferrer, I.; Diaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of β-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Honda, A.; Kawasaki, A.; Nozumi, M. Neuronal Signaling Involved in Neuronal Polarization and Growth: Lipid Rafts and Phosphorylation. Front. Mol. Neurosci. 2020, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.S.; Kumar, S.; Zomorrodi, R.; Ghazala, Z.; Cheam, A.S.M.; Barr, M.S.; Daskalakis, Z.J.; Blumberger, D.M.; Fischer, C.; Flint, A.; et al. Theta-Gamma Coupling and Working Memory in Alzheimer’s Dementia and Mild Cognitive Impairment. Front. Aging Neurosci. 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.-G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Aβ overproduction in a mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Gu, N.; Manseau, F.; Williams, S. Alzheimer’s Transgenic Model Is Characterized by Very Early Brain Network Alterations and β-CTF Fragment Accumulation: Reversal by β-Secretase Inhibition. Front. Cell Neurosci. 2018, 12, 121. [Google Scholar] [CrossRef]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.-P.; et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef]
- Cooper, E.C. Potassium channels (including KCNQ) and epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Xu, Y.; Lavrencic, L.; Radford, K.; Booth, A.; Yoshimura, S.; Anstey, K.J.; Anderson, C.S.; Peters, R. Systematic review of coexistent epileptic seizures and Alzheimer’s disease: Incidence and prevalence. J. Am. Geriatr. Soc. 2021, 69, 2011–2020. [Google Scholar] [CrossRef]
- Manville, R.W.; Abbott, G.W. The Amyloid Precursor Protein C99 Fragment Modulates Voltage-Gated Potassium Channels. Cell Physiol. Biochem. 2021, 55, 157–170. [Google Scholar] [CrossRef]
- Sachse, C.C.; Kim, Y.H.; Agsten, M.; Huth, T.; Alzheimer, C.; Kovacs, D.M.; Kim, D.Y. BACE1 and presenilin/γ-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 2013, 27, 2458–2467. [Google Scholar] [CrossRef] [Green Version]
- Vetrivel, K.S.; Meckler, X.; Chen, Y.; Nguyen, P.D.; Seidah, N.G.; Vassar, R.; Wong, P.C.; Fukata, M.; Kounnas, M.Z.; Thinakaran, G. Alzheimer disease Aβ production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 2009, 284, 3793–3803. [Google Scholar] [CrossRef] [Green Version]
- Dai, G. Neuronal KCNQ2/3 channels are recruited to lipid raft microdomains by palmitoylation of BACE1. J. Gen. Physiol. 2022, 154, e202112888. [Google Scholar] [CrossRef]
- Gallego Villarejo, L.; Bachmann, L.; Marks, D.; Brachthäuser, M.; Geidies, A.; Müller, T. Role of Intracellular Amyloid β as Pathway Modulator, Biomarker, and Therapy Target. Int. J. Mol. Sci. 2022, 23, 4656. [Google Scholar] [CrossRef]
- Tomiyama, T.; Shimada, H. APP Osaka Mutation in Familial Alzheimer’s Disease—Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance. Int. J. Mol. Sci. 2020, 21, 1413. [Google Scholar] [CrossRef] [Green Version]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.Q.; Lannfelt, L.; Nilsson, L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef]
- Kulic, L.; Mcafoose, J.; Welt, T.; Tackenberg, C.; Späni, C.; Wirth, F.; Finder, V.; Konietzko, U.; Giese, M.; Eckert, A.; et al. Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation. Transl. Psychiatry 2012, 2, e183. [Google Scholar] [CrossRef]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Rajendran, L.; Knobloch, M.; Geiger, K.D.; Dienel, S.; Nitsch, R.; Simons, K.; Konietzko, U. Increased Aβ production leads to intracellular accumulation of Aβ in flotillin-1-positive endosomes. Neuro-Degener. Dis. 2007, 4, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef]
- Fleck, D.; Van Bebber, F.; Colombo, A.; Galante, C.; Schwenk, B.M.; Rabe, L.; Hampel, H.; Novak, B.; Kremmer, E.; Tahirovic, S.; et al. Dual Cleavage of Neuregulin 1 Type III by BACE1 and ADAM17 Liberates Its EGF-Like Domain and Allows Paracrine Signaling. J. Neurosci. 2013, 33, 7856–7869. [Google Scholar] [CrossRef] [Green Version]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.C.; Xu, G.; Koliatsos, V.E.; et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef] [Green Version]
- Ben Halima, S.; Mishra, S.; Raja, K.M.P.; Willem, M.; Baici, A.; Simons, K.; Brustle, O.; Koch, P.; Haass, C.; Caflisch, A.; et al. Specific Inhibition of β-Secretase Processing of the Alzheimer Disease Amyloid Precursor Protein. Cell Rep. 2016, 14, 2127–2141. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, L.; Schneider, A.; Schlechtingen, G.; Weidlich, S.; Ries, J.; Braxmeier, T.; Schwille, P.; Schulz, J.B.; Schroeder, C.; Simons, M.; et al. Efficient inhibition of the Alzheimer’s disease β-secretase by membrane targeting. Science 2008, 320, 520–523. [Google Scholar] [CrossRef]
- Houacine, J.; Bolmont, T.; Aeschbach, L.; Oulad-Abdelghani, M.; Fraering, P.C. Selective neutralization of APP-C99 with monoclonal antibodies reduces the production of Alzheimer’s Aβ peptides. Neurobiol. Aging 2012, 33, 2704–2714. [Google Scholar] [CrossRef]
- Paganetti, P.; Calanca, V.; Galli, C.; Stefani, M.; Molinari, M. β-site specific intrabodies to decrease and prevent generation of Alzheimer’s Aβ peptide. J. Cell Biol. 2005, 168, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Matsuda, S.; Giliberto, L.; Arancio, O.; D’Adamio, L. APP heterozygosity averts memory deficit in knockin mice expressing the Danish dementia BRI2 mutant. EMBO J. 2011, 30, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Matsuda, S.; Arancio, O.; D’Adamio, L. β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol. Med. 2012, 4, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, M.; Bicknell, W.; Watson, A.A.; Fairlie, D.P.; Craik, D.J. Solution structure of amyloid β-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry 1998, 37, 11064–11077. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Akcan, M.; Khondker, A.; Rheinstädter, M.C.; Bozelli, J.C.; Epand, R.M.; Huynh, V.; Wylie, R.G.; Boulton, S.; Huang, J.; et al. Atomic resolution map of the soluble amyloid beta assembly toxic surfaces. Chem. Sci. 2019, 10, 6072–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef] [PubMed]
- Mutter, S.T.; Deeth, R.J.; Turner, M.; Platts, J.A. Benchmarking of copper(II) LFMM parameters for studying amyloid-β peptides. J. Biomol. Struct. Dyn. 2018, 36, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali-Torres, J.; Marechal, J.D.; Rodriguez-Santiago, L.; Sodupe, M. Three dimensional models of Cu(2+)-Aβ(1-16) complexes from computational approaches. J. Am. Chem. Soc. 2011, 133, 15008–15014. [Google Scholar] [CrossRef] [PubMed]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Ségalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural Changes of Region 1-16 of the Alzheimer Disease Amyloid β-Peptide upon Zinc Binding and in Vitro Aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozin, S.A.; Zirah, S.; Rebuffat, S.; Hoa, G.H.; Debey, P. Zinc binding to Alzheimer’s Aβ(1-16) peptide results in stable soluble complex. Biochem. Biophys. Res. Commun. 2001, 285, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Colvin, B.A.; Rogers, V.A.; Kulas, J.A.; Ridgway, E.A.; Amtashar, F.S.; Combs, C.K.; Nichols, M.R. The conformational epitope for a new Aβ42 protofibril-selective antibody partially overlaps with the peptide N-terminal region. J. Neurochem. 2017, 143, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Legleiter, J.; Czilli, D.L.; Gitter, B.; DeMattos, R.B.; Holtzman, D.M.; Kowalewski, T. Effect of different anti-Aβ antibodies on Aβ fibrillogenesis as assessed by atomic force microscopy. J. Mol. Biol. 2004, 335, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Racke, M.M.; Boone, L.I.; Hepburn, D.L.; Parsadainian, M.; Bryan, M.T.; Ness, D.K.; Piroozi, K.S.; Jordan, W.H.; Brown, D.D.; Hoffman, W.P.; et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 629–636. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takasugi, N.; Komai, M.; Kaneshiro, N.; Ikeda, A.; Kamikubo, Y.; Uehara, T. The Pursuit of the “Inside” of the Amyloid Hypothesis—Is C99 a Promising Therapeutic Target for Alzheimer’s Disease? Cells 2023, 12, 454. https://doi.org/10.3390/cells12030454
Takasugi N, Komai M, Kaneshiro N, Ikeda A, Kamikubo Y, Uehara T. The Pursuit of the “Inside” of the Amyloid Hypothesis—Is C99 a Promising Therapeutic Target for Alzheimer’s Disease? Cells. 2023; 12(3):454. https://doi.org/10.3390/cells12030454
Chicago/Turabian StyleTakasugi, Nobumasa, Masato Komai, Nanaka Kaneshiro, Atsuya Ikeda, Yuji Kamikubo, and Takashi Uehara. 2023. "The Pursuit of the “Inside” of the Amyloid Hypothesis—Is C99 a Promising Therapeutic Target for Alzheimer’s Disease?" Cells 12, no. 3: 454. https://doi.org/10.3390/cells12030454