

Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses
 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
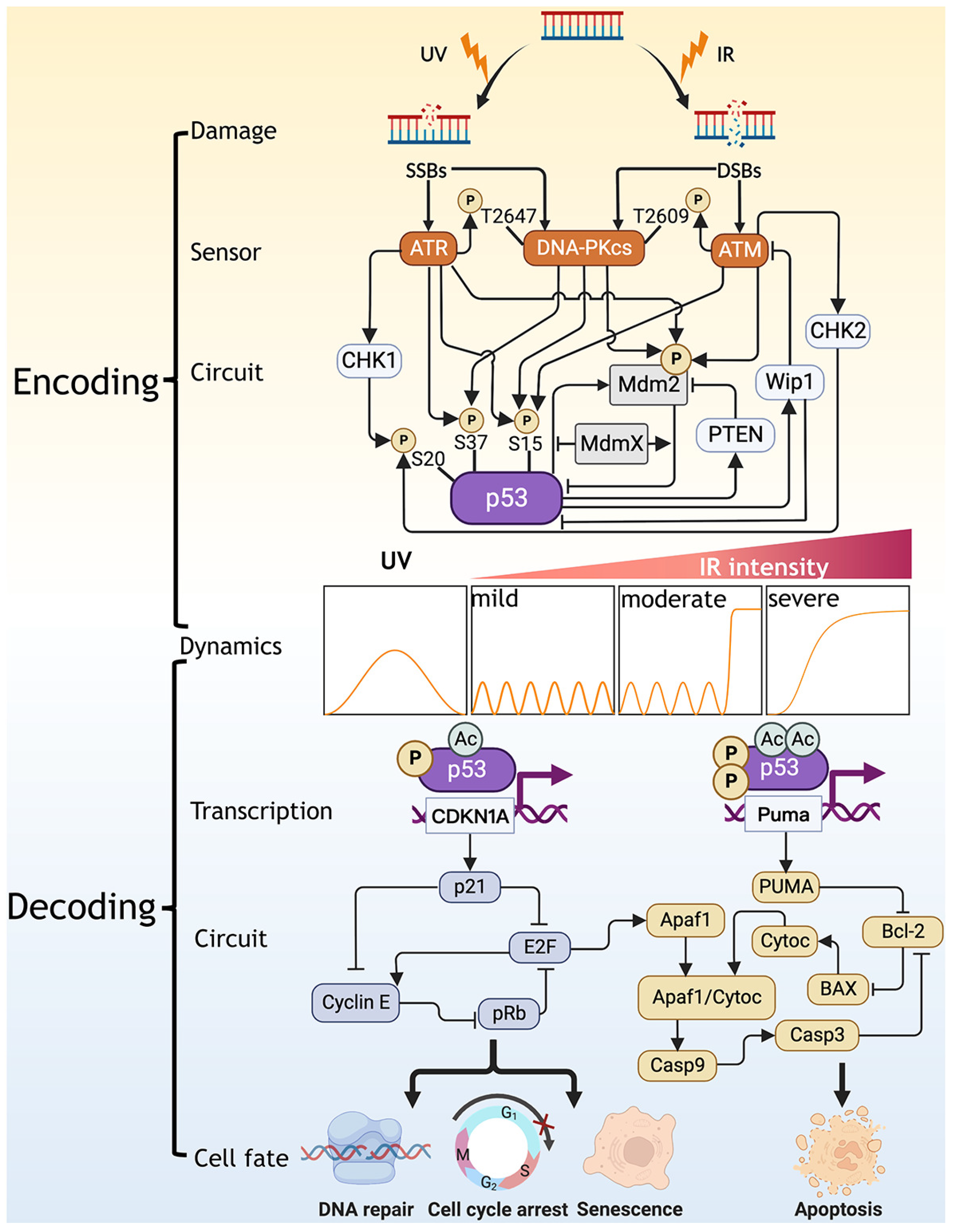
2. Encoding: Translating DNA Damage into p53 Dynamics
2.1. Generation, Sensing, and Repair of DNA Damage
2.2. Stimulus-, Cell Type- and Species-Dependent p53 Dynamics
2.3. Clarifying the Mechanism in the Encoding of p53 Dynamics by Modeling
3. Decoding: Controlling Cell Fate by p53 Dynamics
3.1. p53 Dynamics-Dependent Selection of Target Genes
3.2. Multilevel Modulation of p53 Targets Selection
3.3. Decoding p53 Dynamics into Cell Fate through the Downstream Circuits
4. Mathematical Modeling Provides Insights into a Comprehensive Understanding of p53 Response by Integrating the Encoding and Decoding Processes
4.1. Understanding the Transition between p53 Dynamic Modes
4.2. Coupling p53 Network with Other Signaling Networks in Cell Fate Decisions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 Million Years of Evolution and 40 Years of Discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 Feedback Loop in Individual Cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-Dependent Dynamics of p53 in Single Cells. Mol. Syst. Biol. 2011, 7, 488. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Gan, S.T.; Guan, H.J.; Zhou, Y.; Ouyang, Q.; Shi, J. DNA Damage Strength Modulates a Bimodal Switch of p53 Dynamics for Cell-Fate Control. BMC Biol. 2013, 11, 73. [Google Scholar] [CrossRef]
- Yang, R.; Huang, B.; Zhu, Y.; Li, Y.; Liu, F.; Shi, J. Cell Type-Dependent Bimodal p53 Activation Engenders a Dynamic Mechanism of Chemoresistance. Sci. Adv. 2018, 4, eaat5077. [Google Scholar] [CrossRef]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent Initiation: A Mechanism for Triggering p53 Pulses in Response to DNA Damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Two-Phase Dynamics of p53 in the DNA Damage Response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef]
- Wu, M.; Ye, H.; Tang, Z.; Shao, C.; Lu, G.; Chen, B.; Yang, Y.; Wang, G.; Hao, H. p53 Dynamics Orchestrates with Binding Affinity to Target Genes for Cell Fate Decision. Cell Death Dis. 2017, 8, e3130. [Google Scholar] [CrossRef] [Green Version]
- Caldecott, K.W. DNA Single-Strand Break Repair. Exp. Cell. Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Putnam, C.D.; Tainer, J.A. DNA Double-Strand Break Repair From Head to Tail. Curr. Opin. Struct. Biol. 2002, 12, 115–122. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and Repairing DNA Double-Strand Breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of Non-Homologous End Joining (NHEJ) in Maintaining Genomic Integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Ma, L.; Wagner, J.; Rice, J.J.; Hu, W.; Levine, A.J.; Stolovitzky, G.A. A Plausible Model for the Digital Response of p53 to DNA Damage. Proc. Natl. Acad. Sci. USA 2005, 102, 14266–14271. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Cheng, Z.; Wang, W. Cell Fate Decision Mediated by p53 Pulses. Proc. Natl. Acad. Sci. USA 2009, 106, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Geva-Zatorsky, N.; Rosenfeld, N.; Itzkovitz, S.; Milo, R.; Sigal, A.; Dekel, E.; Yarnitzky, T.; Liron, Y.; Polak, P.; Lahav, G. Oscillations and Variability in the p53 System. Mol. Syst. Biol. 2006, 2, 2006-0033. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA Damage Activates ATM through Intermolecular Autophosphorylation and Dimer Dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Coordination between Cell Cycle Progression and Cell Fate Decision by the p53 and E2F1 Pathways in Response to DNA Damage. J. Biol. Chem. 2010, 285, 31571–31580. [Google Scholar] [CrossRef]
- Loewer, A.; Karanam, K.; Mock, C.; Lahav, G. The p53 Response in Single Cells is Linearly Correlated to the Number of DNA Breaks without a Distinct Threshold. BMC Biol. 2013, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Leal, J.F.M.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-Dependent Phosphorylation of Mdm2 on Serine 395: Role in p53 Activation by DNA Damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, H.M.; Ahmed, S.F.; Sibbet, G.J.; Hristova, V.A.; Nomura, K.; Hock, A.K.; Archibald, L.J.; Jamieson, A.G.; Fushman, D.; Vousden, K.H.; et al. Structural Basis for DNA Damage-Induced Phosphoregulation of MDM2 RING Domain. Nat. Commun. 2020, 11, 2094. [Google Scholar] [CrossRef]
- Shinozaki, T.; Nota, A.; Taya, Y.; Okamoto, K. Functional Role of Mdm2 Phosphorylation by ATR in Attenuation of p53 Nuclear Export. Oncogene 2003, 22, 8870–8880. [Google Scholar] [CrossRef] [PubMed]
- Loewer, A.; Batchelor, E.; Gaglia, G.; Lahav, G. Basal Dynamics of p53 Reveal Transcriptionally Attenuated Pulses in Cycling Cells. Cell 2010, 142, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Hammarsten, O.; DeFazio, L.G.; Chu, G. Activation of DNA-Dependent Protein Kinase by Single-Stranded DNA Ends. J. Biol. Chem. 2000, 275, 1541–1550. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Knudsen, K.E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 2022, 82, 523–533. [Google Scholar] [CrossRef]
- Mayo, L.D.; Turchi, J.J.; Berberich, S.J. Mdm-2 Phosphorylation by DNA-Dependent Protein Kinase Prevents Interaction with p53. Cancer Res. 1997, 57, 5013–5016. [Google Scholar]
- Finzel, A.; Grybowski, A.; Strasen, J.; Cristiano, E.; Loewer, A. Hyperactivation of ATM upon DNA-PKcs Inhibition Modulates p53 Dynamics and Cell Fate in Response to DNA Damage. Mol. Biol. Cell 2016, 27, 2360–2367. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Löbrich, M.; Shiloh, Y.; Chen, D.J. Ataxia Telangiectasia Mutated (ATM) Is Essential for DNA-PKcs Phosphorylations at the Thr-2609 Cluster upon DNA Double Strand Break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef]
- Douglas, P.; Zhong, J.; Ye, R.; Moorhead, G.B.G.; Xu, X.; Lees-Miller, S.P. Protein Phosphatase 6 Interacts with the DNA-dependent Protein Kinase Catalytic Subunit and Dephosphorylates Gamma-H2AX. Mol. Cell. Biol. 2010, 30, 1368–1381. [Google Scholar] [CrossRef]
- Sun, T.; Li, X.; Shen, P. Modeling Amplified p53 Responses under DNA-PK Inhibition in DNA Damage Response. Oncotarget 2017, 8, 17105–17114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev Bar-Or, R.; Maya, R.; Segel, L.A.; Alon, U.; Levine, A.J.; Oren, M. Generation of Oscillations by the p53-Mdm2 Feedback Loop: A Theoretical and Experimental Study. Proc. Natl. Acad. Sci. USA 2000, 97, 11250. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 Dynamics Control Cell Fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, L.; Liu, B.; Liang, X.; Shi, J. Single-Cell Analysis of p53 Transitional Dynamics Unravels Stimulus- and Cell Type-Dependent Signaling Output Motifs. BMC Biol. 2022, 20, 85. [Google Scholar] [CrossRef]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef]
- Chen, S.H.; Forrester, W.; Lahav, G. Schedule-Dependent Interaction between Anticancer Treatments. Science 2016, 351, 1204. [Google Scholar] [CrossRef]
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. p53 Dynamics Vary between Tissues and Are Linked with Radiation Sensitivity. Nat. Commun. 2021, 12, 898. [Google Scholar] [CrossRef]
- Stewart-Ornstein, J.; Cheng, H.W.J.; Lahav, G. Conservation and Divergence of p53 Oscillation Dynamics across Species. Cell Syst. 2017, 5, 410–417.e4. [Google Scholar] [CrossRef]
- Porter, J.R.; Fisher, B.E.; Batchelor, E. p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-Regulated Target Gene Subnetworks. Cell Syst. 2016, 2, 272–282. [Google Scholar] [CrossRef]
- Novák, B.; Tyson, J.J. Design Principles of Biochemical Oscillators. Nat. Rev. Mol. Cell Biol. 2008, 9, 981. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Bao, H.; Zhang, X.P.; Liu, F.; Wang, W. Regulation of Tip60-Dependent p53 Acetylation in Cell Fate Decision. FEBS Lett. 2019, 593, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monke, G.; Cristiano, E.; Finzel, A.; Friedrich, D.; Herzel, H.; Falcke, M.; Loewer, A. Excitability in the p53 Network Mediates Robust Signaling with Tunable Activation Thresholds in Single Cells. Sci. Rep. 2017, 7, 46571. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Ornstein, J.; Lahav, G. p53 Dynamics in Response to DNA Damage Vary across Cell Lines and Are Shaped by Efficiency of DNA Repair and Activity of the Kinase ATM. Sci. Signal. 2017, 10, eaah6671. [Google Scholar] [CrossRef] [PubMed]
- Chong, K.H.; Samarasinghe, S.; Kulasiri, D. Mathematical Modelling of p53 Basal Dynamics and DNA Damage Response. Math. Biosci. 2015, 259, 27–42. [Google Scholar] [CrossRef]
- Kim, J.R.; Yoon, Y.; Cho, K.H. Coupled Feedback Loops Form Dynamic Motifs of Cellular Networks. Biophys. J. 2008, 94, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Brazhnik, P.; Tyson, J.J. Exploring Mechanisms of the DNA-damage Response: p53 Pulses and Their Possible Relevance to Apoptosis. Cell Cycle 2007, 6, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.Y.C.; Choi, Y.S.; Ma, W.; Pomerening, J.R.; Tang, C.; Ferrell, J.E. Robust, Tunable Biological Oscillations from Interlinked Positive and Negative Feedback Loops. Science 2008, 321, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Li, N.X.; Chen, J.J.; Zhang, X.P.; Liu, F.; Wang, W. Modulation of Dynamic Modes by Interplay between Positive and Negative Feedback Loops in Gene Regulatory Networks. Phys. Rev. E 2018, 97, 042412. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Zhang, X.P.; Liu, F.; Wang, W. Involvement of miR-605 and miR-34a in the DNA Damage Response Promotes Apoptosis Induction. Biophys. J. 2014, 106, 1792–1800. [Google Scholar] [CrossRef]
- Zhang, X.P.; Liu, F.; Wang, W. Interplay between Mdm2 and HIPK2 in the DNA Damage Response. J. R. Soc. Interface 2014, 11, 20140319. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.J.; Liu, F.; Zhang, X.P.; Li, J.; Wang, W. A Two-Step Mechanism for Cell Fate Decision by Coordination of Nuclear and Mitochondrial p53 Activities. PLoS ONE 2012, 7, e38164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuge, C.; Sun, X.; Chen, Y.; Lei, J. PDCD5 Functions as a Regulator of p53 Dynamics in the DNA Damage Response. J. Theor. Biol. 2016, 388, 1–10. [Google Scholar] [CrossRef]
- Sun, C.Y.; Zhang, X.P.; Wang, W. Coordination of miR-192 and miR-22 in P53-Mediated Cell Fate Decision. Int. J. Mol. Sci. 2019, 20, 4768. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.; Ooi, H.K.; Kang, T.; Bleris, L.; Ma, L. MiR-192-Mediated Positive Feedback Loop Controls the Robustness of Stress-Induced p53 Oscillations in Breast Cancer Cells. PLoS Comput. Biol. 2015, 11, e1004653. [Google Scholar] [CrossRef]
- Suarez, O.; Vega, C.J.; Sanchez, E.; Gonzalez-Santiago, A. Cell Death Induction in P53-Mdm2 Network Regulated by P300 and HDAC1 Using Pinning Control. In Proceedings of the 2019 16th International Conference on Electrical Engineering, Computing Science and Automatic Control (CCE), Mexico City, Mexico, 11–13 September 2019; pp. 1–6. [Google Scholar]
- Suarez, O.J.; Vega, C.J.; Sanchez, E.N.; González-Santiago, A.E.; Rodríguez-Jorge, O.; Alanis, A.Y.; Chen, G.; Hernandez-Vargas, E.A. Pinning Control for the p53-Mdm2 Network Dynamics Regulated by p14ARF. Front. Physiol. 2020, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-Driven Experimental Approach Reveals the Complex Regulatory Distribution of p53 by the Circadian Factor Period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A Global Map of p53 Transcription-Factor Binding Sites in the Human Genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A Threshold Mechanism Mediates p53 Cell Fate Decision Between Growth Arrest and Apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A Complex Barcode underlies the Heterogeneous Response of p53 to Stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Harton, M.D.; Koh, W.S.; Bunker, A.D.; Singh, A.; Batchelor, E. p53 Pulse Modulation Differentially Regulates Target Gene Promoters to Regulate Cell Fate Decisions. Mol. Syst. Biol. 2019, 15, e8685. [Google Scholar] [CrossRef]
- Hafner, A.; Reyes, J.; Stewart-Ornstein, J.; Tsabar, M.; Jambhekar, A.; Lahav, G. Quantifying the Central Dogma in the p53 Pathway in Live Single Cells. Cell Syst. 2020, 10, 495–505.e4. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Stewart-Ornstein, J.; Purvis, J.E.; Forrester, W.C.; Bulyk, M.L.; Lahav, G. p53 Pulses Lead to Distinct Patterns of Gene Expression Albeit Similar DNA-binding Dynamics. Nat. Struct. Mol. Biol. 2017, 24, 840–847. [Google Scholar] [CrossRef]
- Hanson, R.L.; Porter, J.R.; Batchelor, E. Protein Stability of p53 Targets Determines Their Temporal Expression Dynamics in Response to p53 Pulsing. J. Cell Biol. 2019, 218, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, A.; Lu, D.; Kalocsay, M.; Berberich, M.J.; Balbi, P.; Jambhekar, A.; Lahav, G. Time-series Transcriptomics and Proteomics Reveal Alternative Modes to Decode p53 Oscillations. Mol. Syst. Biol. 2022, 18, e10588. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate p53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and Control of p53 Tetramerization in Individual Living Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501. [Google Scholar] [CrossRef]
- Friedrich, D.; Friedel, L.; Finzel, A.; Herrmann, A.; Preibisch, S.; Loewer, A. Stochastic Transcription in the p53-mediated Response to DNA Damage Is Modulated by Burst Frequency. Mol. Syst. Biol. 2019, 15, e9068. [Google Scholar] [CrossRef] [PubMed]
- Friedel, L.; Loewer, A. The Guardian’s Choice: How p53 Enables Context-Specific Decision-Making in Individual Cells. FEBS J. 2022, 289, 40–52. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent Acetylation of p53 Modulates the Decision Between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 Determines the Induction of PUMA and Apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.S.; Kayed, R.; Abate, G.; Uberti, D.; Kinnon, P.; Piccirella, S. Post-Translational Modifications of the p53 Protein and the Impact in Alzheimer’s Disease: A Review of the Literature. Front. Aging Neurosci. 2022, 14, 835288. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Gu, B.; Zhu, W.G. Surf the Post-Translational Modification Network of p53 Regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [PubMed]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of p53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice Lacking p21CIP1/WAF1 Undergo Normal Development, but Are Defective in G1 Checkpoint Control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and Cell Death: Outer Membrane Permeabilization and Beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Ow, Y.L.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond Respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Bose, I.; Ghosh, B. The p53-MDM2 Network: From Oscillations to Apoptosis. J. Biosci. 2007, 32, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Wang, W. Regulation of the DNA Damage Response by p53 Cofactors. Biophys. J. 2012, 102, 2251–2260. [Google Scholar] [CrossRef] [Green Version]
- Pu, T.; Zhang, X.P.; Liu, F.; Wang, W. Coordination of the Nuclear and Cytoplasmic Activities of p53 in Response to DNA Damage. Biophys. J. 2010, 99, 1696–1705. [Google Scholar] [CrossRef]
- Bagci, E.Z.; Vodovotz, Y.; Billiar, T.R.; Ermentrout, G.B.; Bahar, I. Bistability in Apoptosis: Roles of Bax, Bcl-2, and Mitochondrial Permeability Transition Pores. Biophys. J. 2006, 90, 1546–1559. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Towards DNA-damage Induced Autophagy: A Boolean Model of P53-Induced Cell Fate Mechanisms. DNA Repair 2020, 96, 102971. [Google Scholar] [CrossRef]
- Salomao, N.; Karakostis, K.; Hupp, T.; Vollrath, F.; Vojtesek, B.; Fahraeus, R. What Do We Need to Know and Understand about p53 to Improve Its Clinical Value? J. Pathol. 2021, 254, 443–453. [Google Scholar] [CrossRef] [PubMed]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of Wild-Type p53 by Hypoxia-Inducible Factor 1 Alpha. Nature 1998, 392, 405–408. [Google Scholar] [CrossRef]
- Lee, S.J.; Lim, C.J.; Min, J.K.; Lee, J.K.; Kim, Y.M.; Lee, J.Y.; Won, M.H.; Kwon, Y.G. Protein Phosphatase 1 Nuclear Targeting Subunit is A Hypoxia Inducible Gene: Its Role in Post-Translational Modification of p53 and MDM2. Cell Death Differ. 2007, 14, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Polansky, H.; Schwab, H. How A Disruption of the Competition between HIF-1 and p53 for Limiting P300/CBP by Latent Viruses can Cause Disease. Genes Cancer 2018, 9, 153–154. [Google Scholar] [CrossRef]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 Modulates both Apoptosis and Radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef]
- Zhou, C.H.; Zhang, X.P.; Liu, F.; Wang, W. Modeling the Interplay between the HIF-1 and p53 Pathways in Hypoxia. Sci. Rep. 2015, 5, 13834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.W.; Zhang, X.P.; Liu, F.; Ye, X.W.; Zhang, X.P.; Liu, F. CSB Modulates the Competition between HIF-1 and p53 upon Hypoxia. Math. Biosci. Eng. 2019, 16, 5262–5274. [Google Scholar] [CrossRef]
- Itahana, K.; Pervaiz, S. Crosstalk Between p53 and Mitochondrial Metabolism. In Mitochondria: The Anti-Cancer Target for the Third Millennium; Neuzil, J., Pervaiz, S., Fulda, S., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 327–348. [Google Scholar]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, Myc and HIF-1 in Regulating Glycolysis — the Seventh Hallmark of Cancer. Cell. Mol. Life Sci. 2008, 65, 3981. [Google Scholar] [CrossRef]
- Green, D.R.; Chipuk, J.E. p53 and Metabolism: Inside the TIGAR. Cell 2006, 126, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Konrath, F.; Mittermeier, A.; Cristiano, E.; Wolf, J.; Loewer, A. A Systematic Approach to Decipher Crosstalk in the p53 Signaling Pathway Using Single Cell Dynamics. PLoS Comput. Biol. 2020, 16, e1007901. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in The Cross Fire: p53 in Inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Heyne, K.; Winter, C.; Gerten, F.; Schmidt, C.; Roemer, K. A Novel Mechanism of Crosstalk Between the p53 and NF-κB Pathways: MDM2 Binds and Inhibits P65RelA. Cell Cycle 2013, 12, 2479–2492. [Google Scholar] [CrossRef]
- Ikeda, A.; Sun, X.; Li, Y.; Zhang, Y.; Eckner, R.; Doi, T.S.; Takahashi, T.; Obata, Y.; Yoshioka, K.; Yamamoto, K. P300/CBP-dependent and -independent Transcriptional Interference between NF-κB RelA and p53. Biochem. Biophys. Res. Commun. 2000, 272, 375–379. [Google Scholar] [CrossRef]
- Ohtani, K.; DeGregori, J.; Nevins, J.R. Regulation of the Cyclin E Gene by Tanscription Factor E2F1. Proc. Natl. Acad. Sci. USA 1995, 92, 12146–12150. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in Life and Death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int. J. Mol. Sci. 2017, 18, 2537. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, J.J. The Mdm2-p53 Relationship Evolves: Mdm2 Swings both Ways as an Oncogene and A Tumor Suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef]
- Malmlöf, M.; Roudier, E.; Högberg, J.; Stenius, U. MEK-ERK-Mediated Phosphorylation of Mdm2 at Ser-166 in Hepatocytes: Mdm2 is Activated in Response to Inhibited Akt Signaling. J. Biol. Chem. 2007, 282, 2288–2296. [Google Scholar] [CrossRef]
- Araki, S.; Eitel, J.A.; Batuello, C.N.; Bijangi-Vishehsaraei, K.; Xie, X.J.; Danielpour, D.; Pollok, K.E.; Boothman, D.A.; Mayo, L.D. TGF-β1–Induced Expression of Human Mdm2 Correlates with Late-Stage Metastatic Breast Cancer. J. Clin. Investig. 2010, 120, 290–302. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.J.; Zhang, H.; Xing, J. Coupled Reversible and Irreversible Bistable Switches Underlying TGFβ-induced Epithelial to Mesenchymal Transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic Roles of EMT-inducing Transcription Factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Xing, J.; Tian, X.J. Investigating Epithelial-to-Mesenchymal Transition with Integrated Computational and Experimental Approaches. Phys. Biol. 2019, 16, 031001. [Google Scholar] [CrossRef]
- Batchelor, E.; Loewer, A.; Lahav, G. The Ups and Downs of p53: Understanding Protein Dynamics in Single Cells. Nat. Rev. Cancer 2009, 9, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Chen, J.Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubbutat, M.H.; Vousden, K.H. Proteolytic Cleavage of Human p53 by Calpain: A Potential Regulator of Protein Stability. Mol. Cell. Biol. 1997, 17, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Sedarous, M.; Keramaris, E.; O’Hare, M.; Melloni, E.; Slack, R.S.; Elce, J.S.; Greer, P.A.; Park, D.S. Calpains Mediate p53 Activation and Neuronal Death Evoked by DNA Damage *. J. Biol. Chem. 2003, 278, 26031–26038. [Google Scholar] [CrossRef]
- Jentsch, M.; Snyder, P.; Sheng, C.; Cristiano, E.; Loewer, A. p53 Dynamics in Single Cells Are Temperature-Sensitive. Sci. Rep. 2020, 10, 1481. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Wang, H.-Y.; Gao, X.-J.; Zhu, H.-X.; Zhang, X.-P.; Liu, F.; Wang, W. Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses. Cells 2023, 12, 490. https://doi.org/10.3390/cells12030490
Wang P, Wang H-Y, Gao X-J, Zhu H-X, Zhang X-P, Liu F, Wang W. Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses. Cells. 2023; 12(3):490. https://doi.org/10.3390/cells12030490
Chicago/Turabian StyleWang, Ping, Hang-Yu Wang, Xing-Jie Gao, Hua-Xia Zhu, Xiao-Peng Zhang, Feng Liu, and Wei Wang. 2023. "Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses" Cells 12, no. 3: 490. https://doi.org/10.3390/cells12030490
APA StyleWang, P., Wang, H. -Y., Gao, X. -J., Zhu, H. -X., Zhang, X. -P., Liu, F., & Wang, W. (2023). Encoding and Decoding of p53 Dynamics in Cellular Response to Stresses. Cells, 12(3), 490. https://doi.org/10.3390/cells12030490