
Protective Effect of NO2-OA on Oxidative Stress, Gliosis, and Pro-Angiogenic Response in Müller Glial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Line and Culture Reagents
2.3. Cell Viability Assay
2.4. Western Blot Assay
2.5. Dichlorofluorescein Assay
2.6. Quantitative Real-Time Reverse-Transcription PCR (qRT-PCR)
2.7. Hypoxic Assays
2.8. Tube Formation Assay
2.9. Statistical Analysis
3. Results
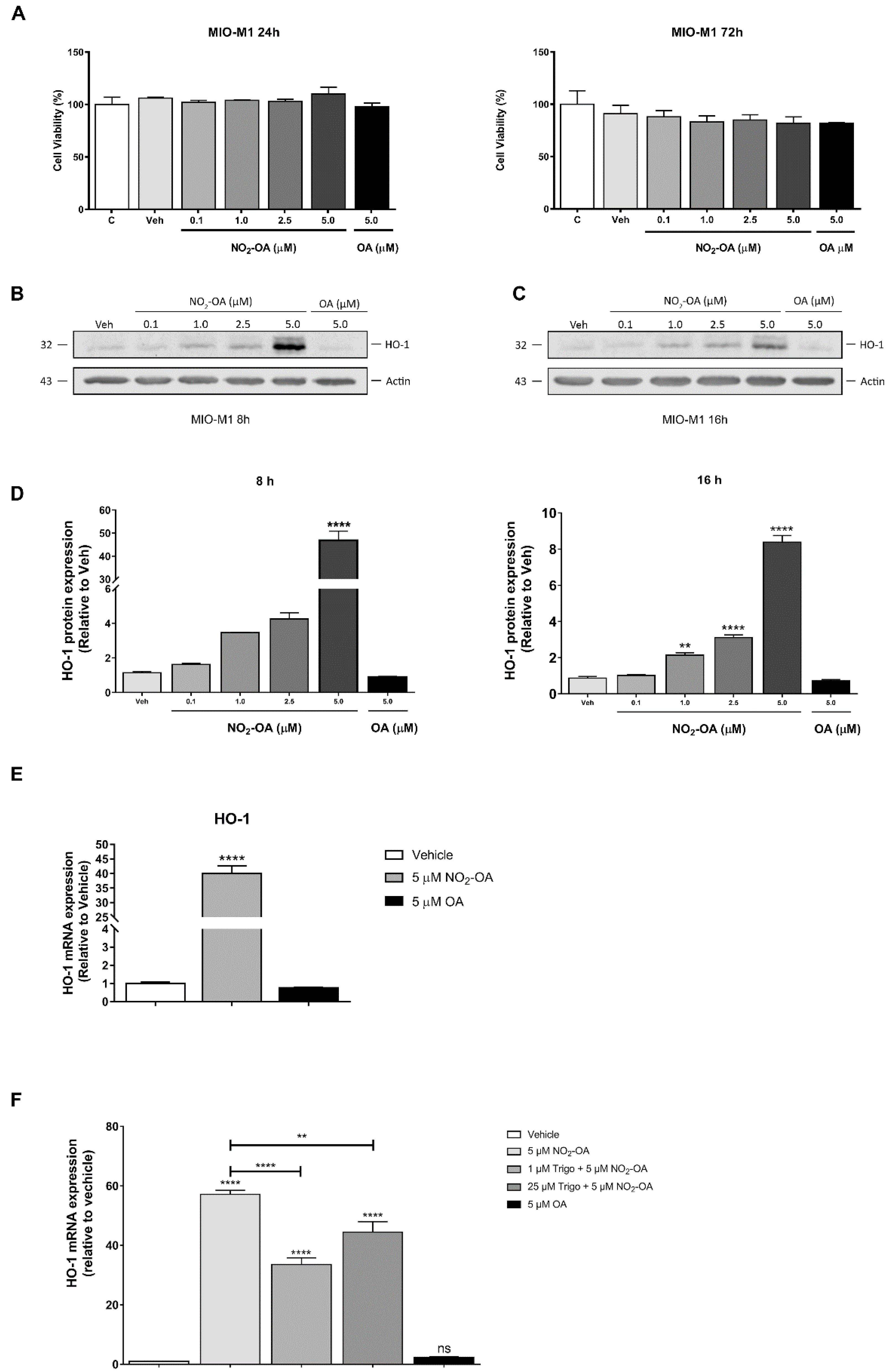
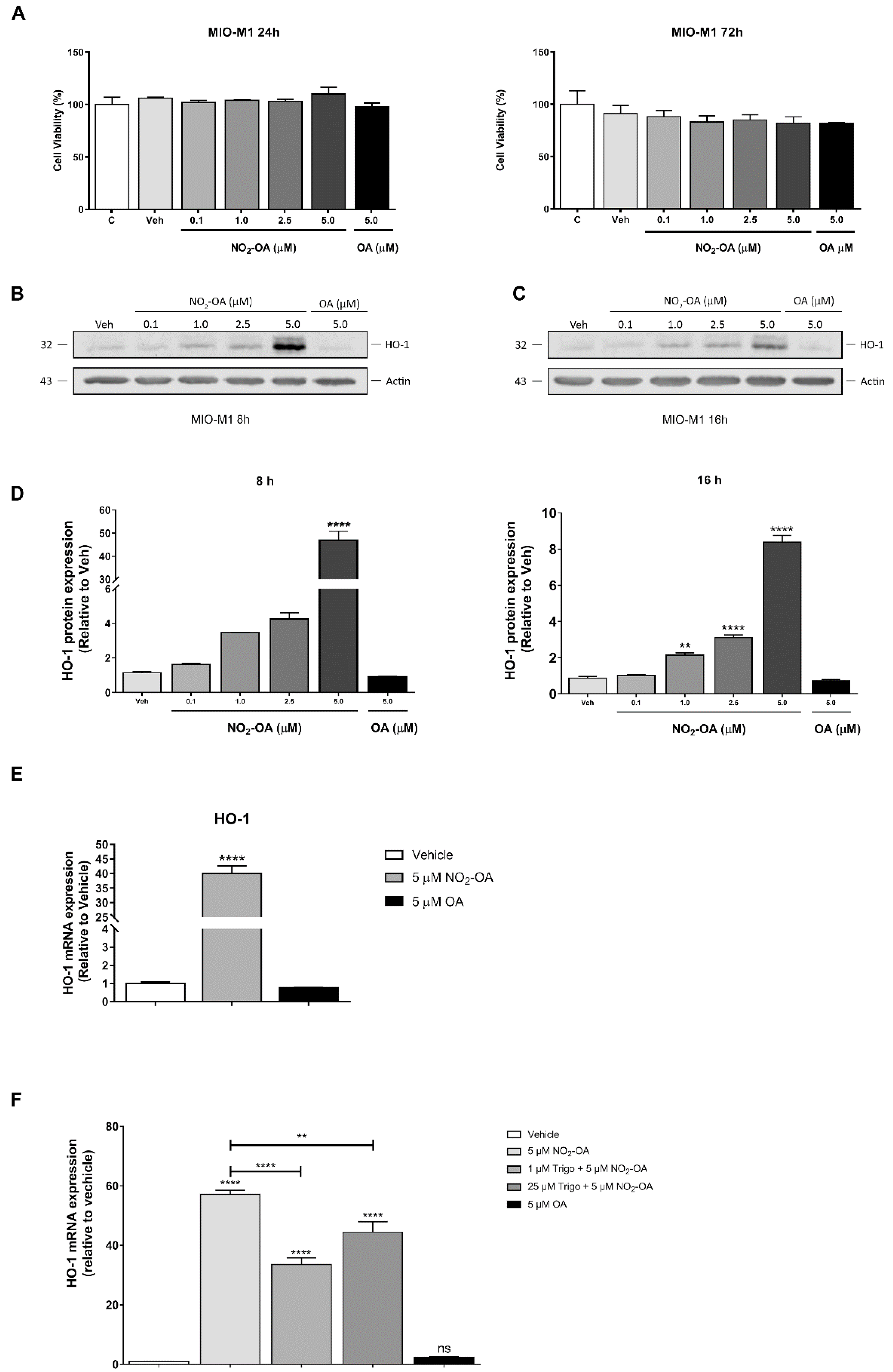
3.1. NO2-OA Induces HO-1 Expression in MGCs
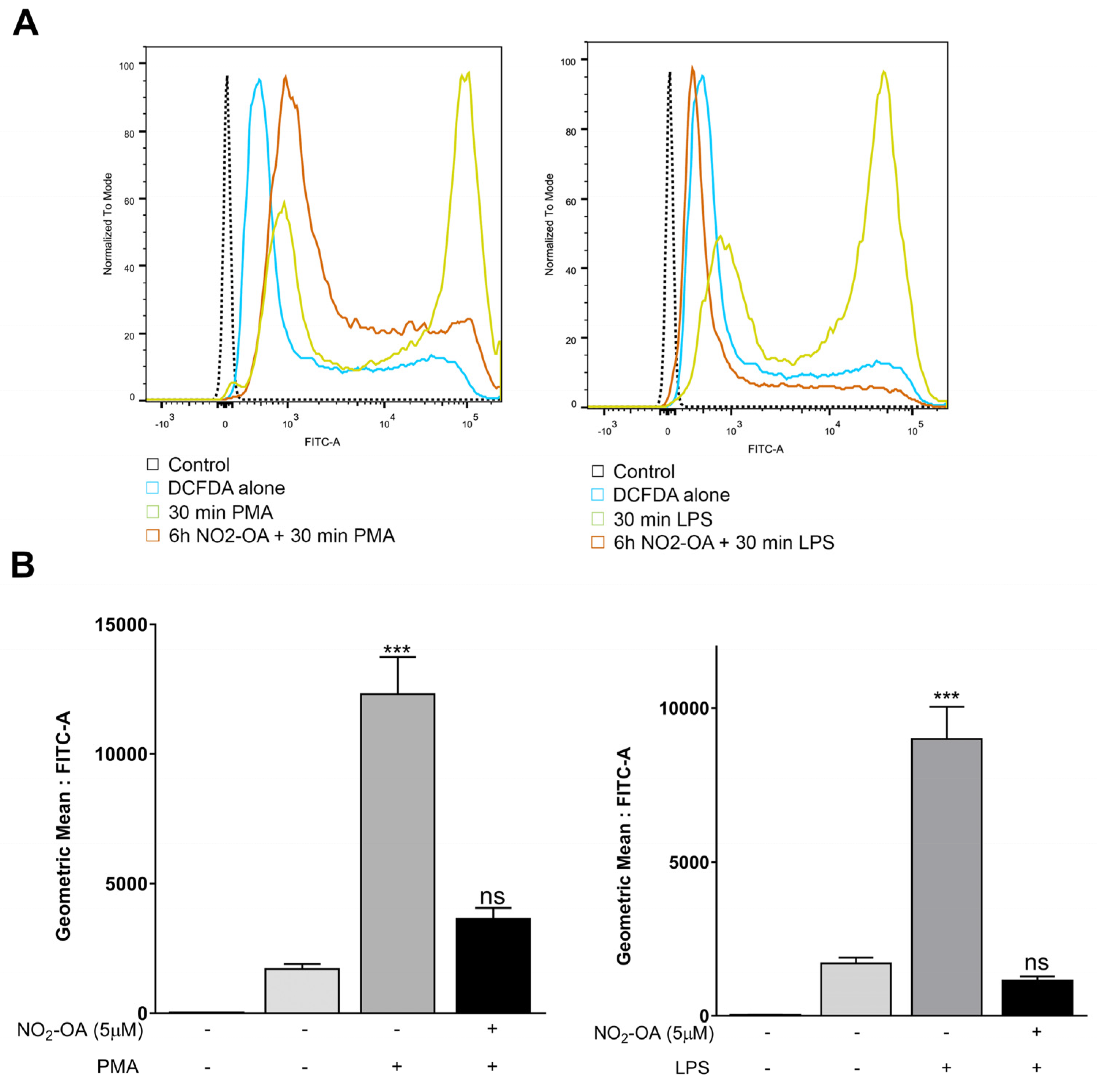
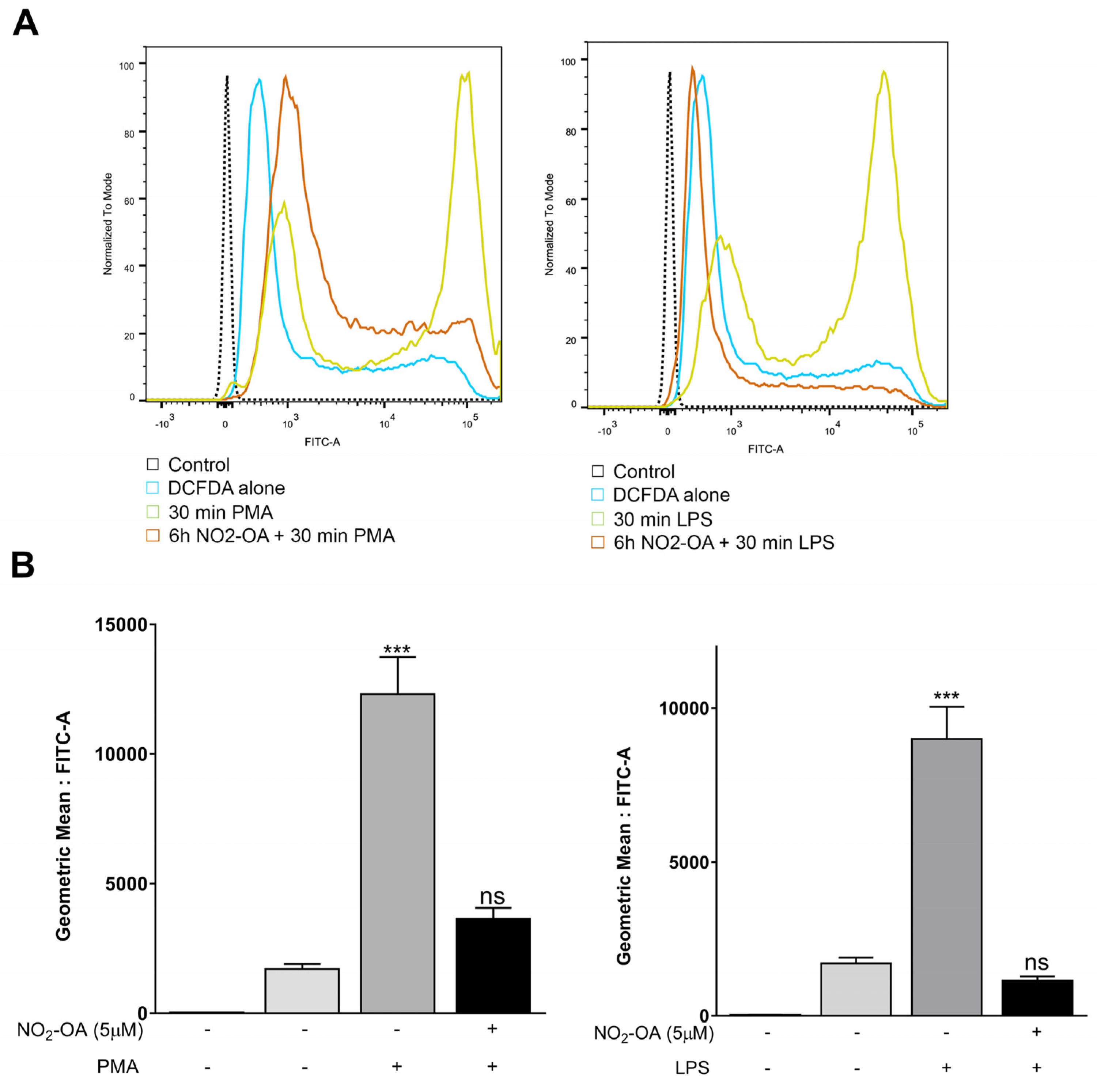
3.2. NO2-OA Prevents the Increase in ROS Levels Induced by LPS or PMA in MGCs
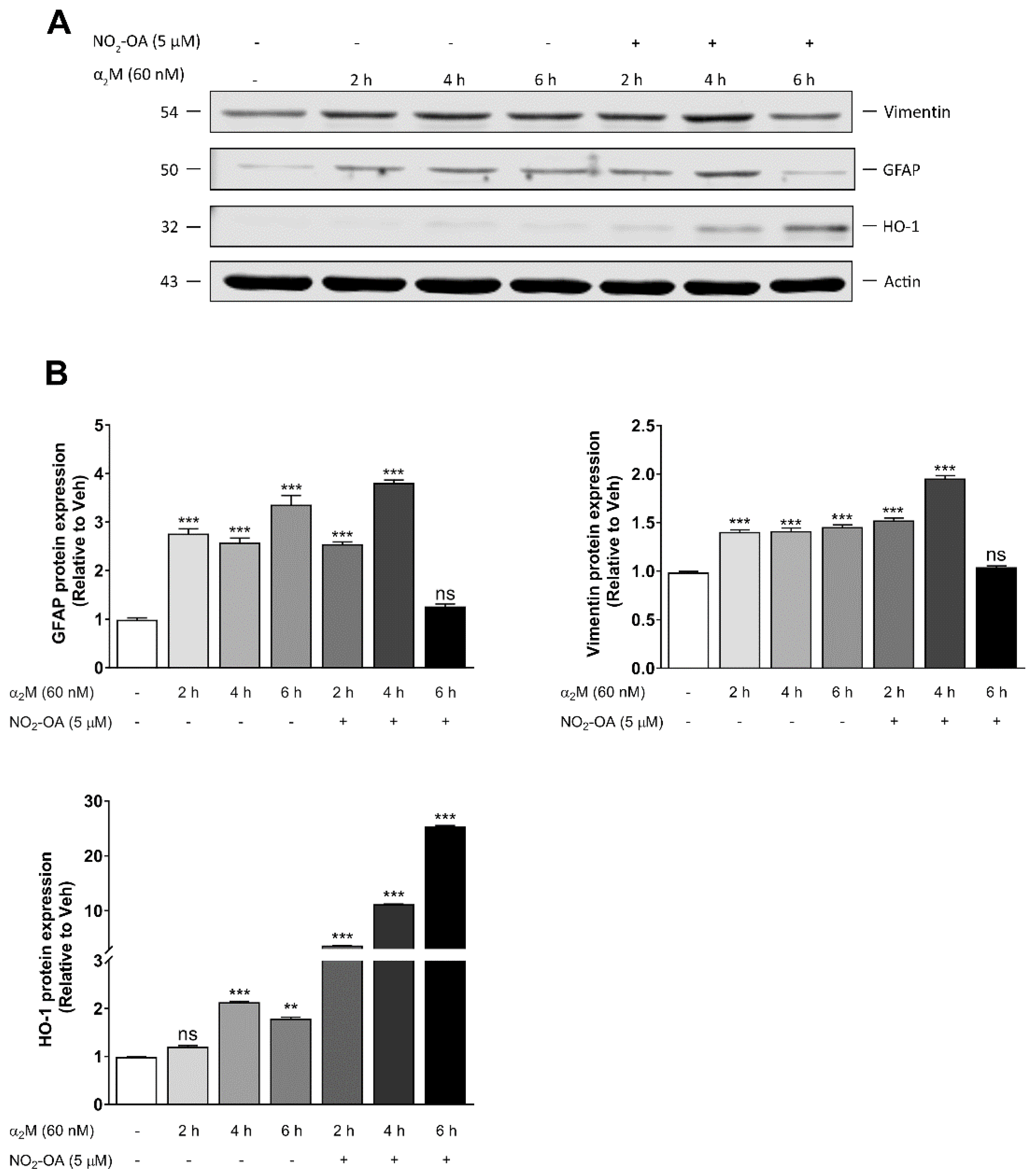
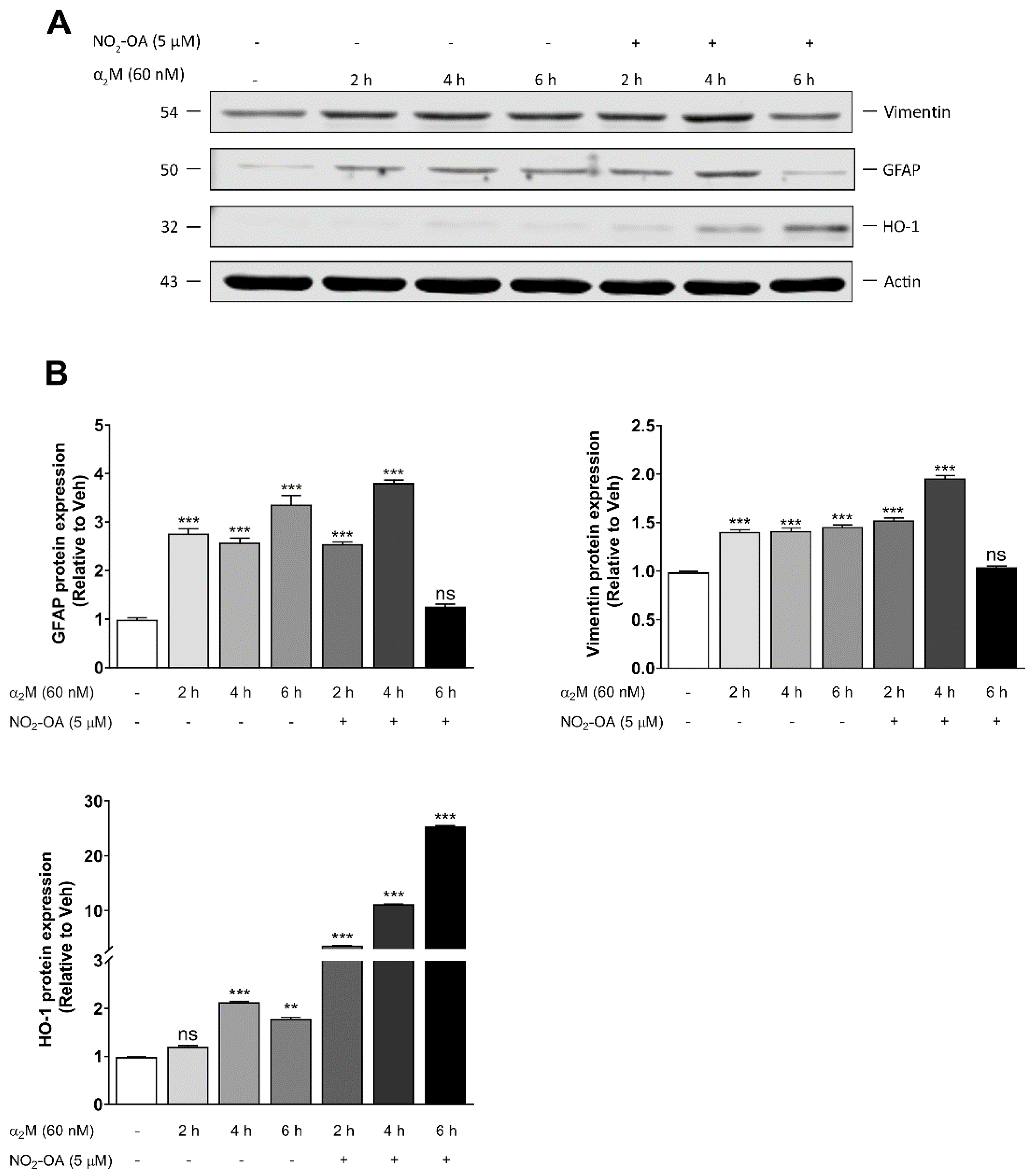
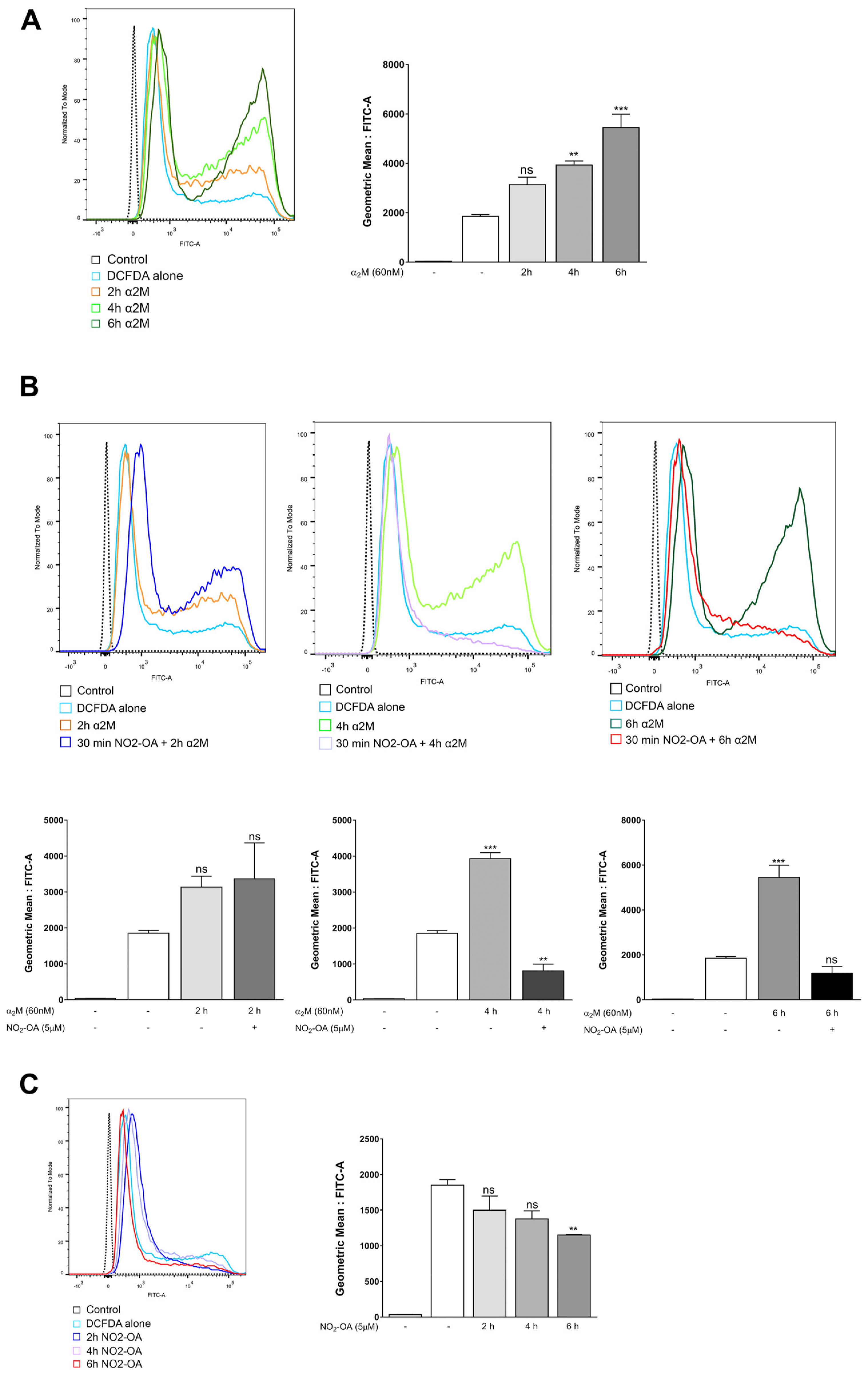
3.3. NO2-OA Reverts Glial Stress Induced by α2M* in MGCs
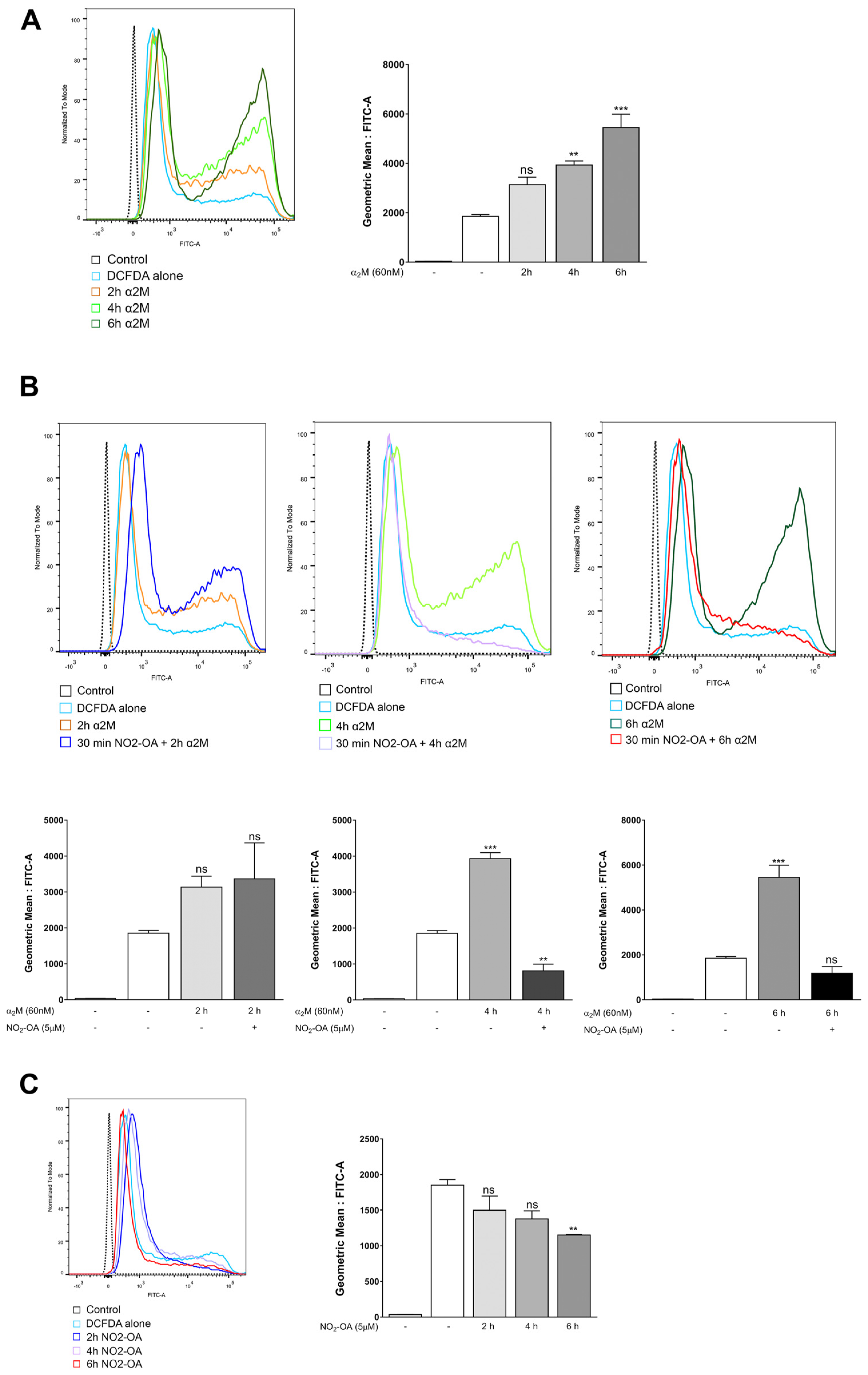
3.4. NO2-OA Inhibits ROS Induced by α2M* in MGCs
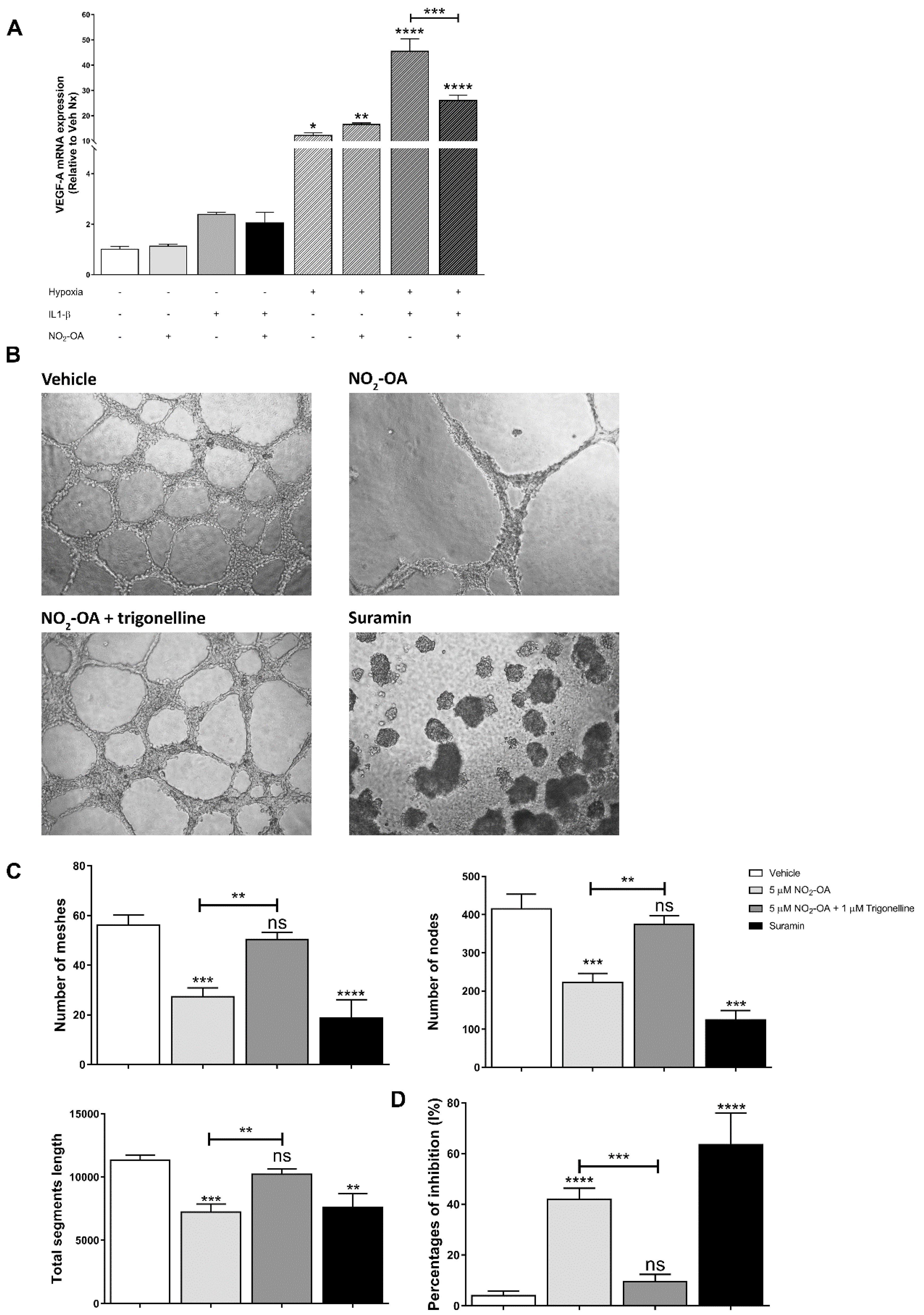
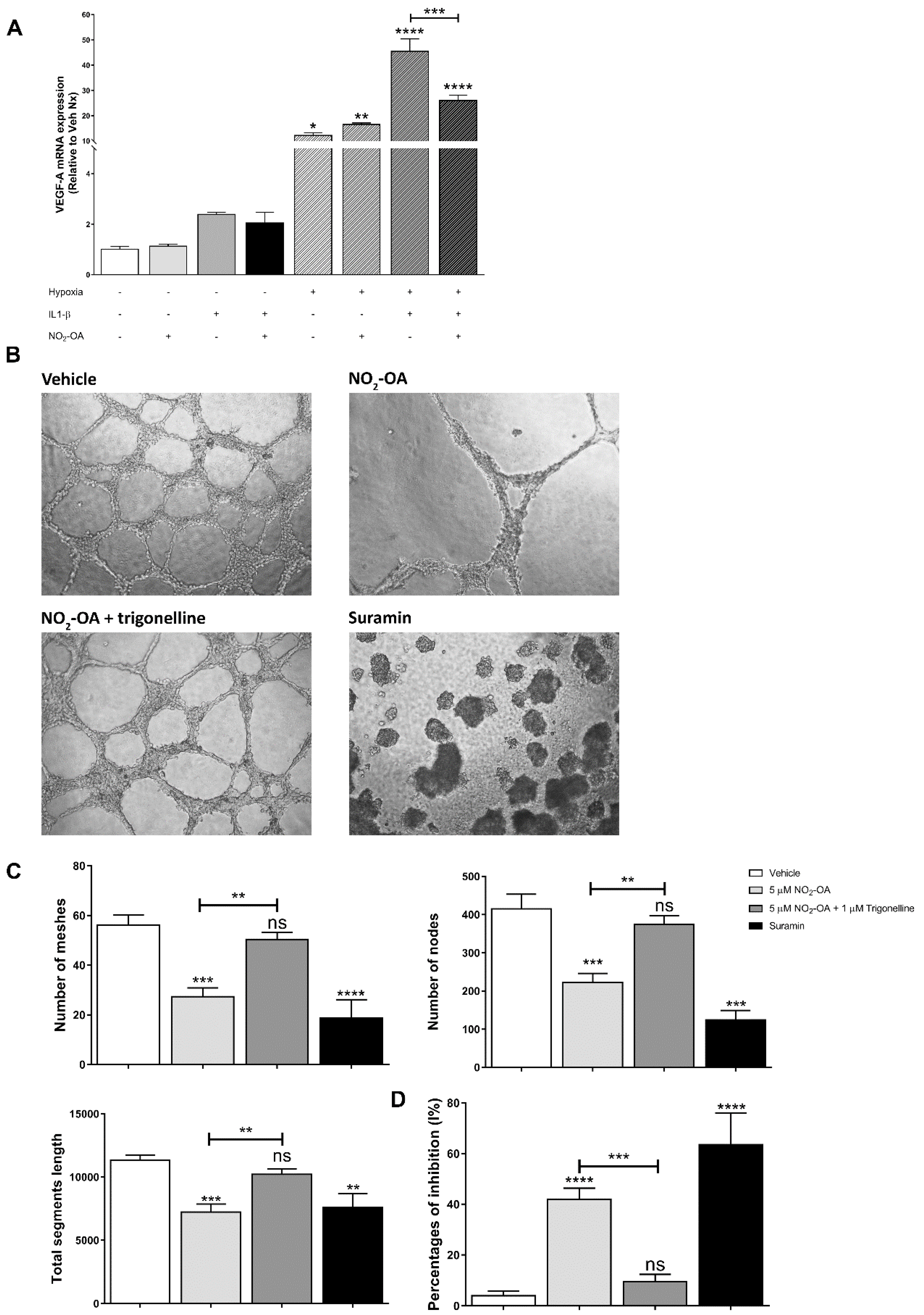
3.5. Effect of NO2-OA on Angiogenesis in MGCs and ECs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedlander, M.; Dorrell, M.I.; Ritter, M.R.; Marchetti, V.; Moreno, S.K.; El-Kalay, M.; Bird, A.C.; Banin, E.; Aguilar, E. Progenitor cells and retinal angiogenesis. Angiogenesis 2007, 10, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Durham, J.T.; Herman, I.M. Microvascular modifications in diabetic retinopathy. Curr. Diab. Rep. 2011, 11, 253–264. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell. Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Rodrigues, M.; Xin, X.; Jee, K.; Babapoor-Farrokhran, S.; Kashiwabuchi, F.; Ma, T.; Bhutto, I.; Hassan, S.J.; Daoud, Y.; Baranano, D.; et al. VEGF secreted by hypoxic Müller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes 2013, 62, 3863–3873. [Google Scholar] [CrossRef]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Graca, A.B.; Hippert, C.; Pearson, R.A. Müller Glia Reactivity and Development of Gliosis in Response to Pathological Conditions. Adv. Exp. Med. Biol. 2018, 1074, 303–308. [Google Scholar]
- Liu, Q.; Ling, T.Y.; Shieh, H.S.; Johnson, F.E.; Huang, J.S.; Huang, S.S. Identification of the high affinity binding site in transforming growth factor-beta involved in complex formation with alpha 2-macroglobulin. Implications regarding the molecular mechanisms of complex formation between alpha 2-macroglobulin and growth factors, cytokines, and hormones. J. Biol. Chem. 2001, 276, 46212–46218. [Google Scholar] [PubMed]
- Garcia-Ferrer, I.; Marrero, A.; Gomis-Rüth, F.X.; Goulas, T. α 2-Macroglobulins: Structure and Function. Subcell. Biochem. 2017, 83, 149–183. [Google Scholar] [PubMed]
- Barcelona, P.F.; Ortiz, S.G.; Chiabrando, G.A.; Sánchez, M.C. alpha2-Macroglobulin induces glial fibrillary acidic protein expression mediated by low-density lipoprotein receptor-related protein 1 in Müller cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Müller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Hoh, J.; Liu, J. Müller cells in pathological retinal angiogenesis. Transl. Res. 2019, 207, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Sapieha, P.; Joyal, J.S.; Rivera, J.C.; Kermorvant-Duchemin, E.; Sennlaub, F.; Hardy, P.; Lachapelle, P.; Chemtob, S. Retinopathy of prematurity: Understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Investig. 2010, 120, 3022–3032. [Google Scholar] [CrossRef]
- Bonacci, G.; Baker, P.R.S.; Salvatore, S.R.; Shores, D.; Khoo, N.K.H.; Koenitzer, J.R.; Vitturi, D.A.; Woodcock, S.R.; Golin-Bisello, F.; Cole, M.P.; et al. Conjugated linoleic acid is a preferential substrate for fatty acid nitration. J. Biol. Chem. 2012, 287, 44071–44082. [Google Scholar] [CrossRef] [PubMed]
- Vitturi, D.A.; Minarrieta, L.; Salvatore, S.R.; Postlethwait, E.M.; Fazzari, M.; Ferrer-Sueta, G.; Lancaster, J.R.; Freeman, B.A.; Schopfer, F.J. Convergence of biological nitration and nitrosation via symmetrical nitrous anhydride. Nat. Chem. Biol. 2015, 11, 504–510. [Google Scholar] [CrossRef]
- Villacorta, L.; Minarrieta, L.; Salvatore, S.R.; Khoo, N.K.; Rom, O.; Gao, Z.; Berman, R.C.; Jobbagy, S.; Li, L.; Woodcock, S.R.; et al. In situ generation, metabolism and immunomodulatory signaling actions of nitro-conjugated linoleic acid in a murine model of inflammation. Redox Biol. 2018, 15, 522–531. [Google Scholar] [CrossRef]
- Tsikas, D.; Zoerner, A.A.; Mitschke, A.; Gutzki, F.M. Nitro-fatty acids occur in human plasma in the picomolar range: A targeted nitro-lipidomics GC-MS/MS study. Lipids 2009, 44, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, S.R.; Rowart, P.; Schopfer, F.J. Mass spectrometry-based study defines the human urine nitrolipidome. Free Radic. Biol. Med. 2021, 162, 327–337. [Google Scholar] [CrossRef]
- Rudolph, V.; Rudolph, T.K.; Schopfer, F.J.; Bonacci, G.; Woodcock, S.R.; Cole, M.P.; Baker, P.R.S.; Ramani, R.; Freeman, B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010, 85, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Koenitzer, J.R.; Bonacci, G.; Woodcock, S.R.; Chen, C.S.; Cantu-Medellin, N.; Kelley, E.E.; Schopfer, F.J. Fatty acid nitroalkenes induce resistance to ischemic cardiac injury by modulating mitochondrial respiration at complex II. Redox. Biol. 2016, 8, 1–10. [Google Scholar] [CrossRef]
- Turell, L.; Vitturi, D.A.; Coitiño, E.L.; Lebrato, L.; Möller, M.N.; Sagasti, C.; Salvatore, S.R.; Woodcock, S.R.; Alvarez, B.; Schopfer, F.J. The Chemical Basis of Thiol Addition to Nitro-conjugated Linoleic Acid, a Protective Cell-signaling Lipid. J. Biol. Chem. 2017, 292, 1145–1159. [Google Scholar] [CrossRef]
- Trostchansky, A.; Bonilla, L.; González-Perilli, L.; Rubbo, H. Nitro-fatty acids: Formation, redox signaling, and therapeutic potential. Antioxid. Redox Signal. 2013, 19, 1257–1265. [Google Scholar] [CrossRef]
- Diaz-Amarilla, P.; Miquel, E.; Trostchansky, A.; Trias, E.; Ferreira, A.M.; Freeman, B.A.; Cassina, P.; Barbeito, L.; Vargas, M.R.; Rubbo, H. Electrophilic nitro-fatty acids prevent astrocyte-mediated toxicity to motor neurons in a cell model of familial amyotrophic lateral sclerosis via nuclear factor erythroid 2-related factor activation. Free Radic. Biol. Med. 2016, 95, 112–120. [Google Scholar] [CrossRef]
- Nakamura, S.; Noguchi, T.; Inoue, Y.; Sakurai, S.; Nishinaka, A.; Hida, Y.; Masuda, T.; Nakagami, Y.; Horai, N.; Tsusaki, H.; et al. Nrf2 Activator RS9 Suppresses Pathological Ocular Angiogenesis and Hyperpermeability. Invest. Ophthalmol. Vis. Sci. 2019, 60, 1943–1952. [Google Scholar] [CrossRef]
- Huang, Z.; Kin Ng, T.; Chen, W.; Sun, X.; Huang, D.; Zheng, D.; Yi, J.; Xu, Y.; Zhuang, X.; Chen, S. Nattokinase Attenuates Retinal Neovascularization Via Modulation of Nrf2/HO-1 and Glial Activation. Investig. Ophthalmol. Vis. Sci. 2021, 62, 25. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gong, J.; Xu, Z.; Duh, E.J. Nrf2 promotes reparative angiogenesis through regulation of NADPH oxidase-2 in oxygen-induced retinopathy. Free Radic. Biol. Med. 2016, 99, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef]
- Chiabrando, G.; Bonacci, G.; Sanchez, C.; Ramos, A.; Zalazar, F.; Vides, M.A. A procedure for human pregnancy zone protein (and human alpha 2-macroglobulin) purification using hydrophobic interaction chromatography on phenyl-sepharose CL-4B column. Protein. Expr. Purif. 1997, 9, 399–406. [Google Scholar] [CrossRef]
- Chiabrando, G.A.; Sánchez, M.C.; Skornicka, E.L.; Koo, P.H. Low-density lipoprotein receptor-related protein mediates in PC12 cell cultures the inhibition of nerve growth factor-promoted neurite outgrowth by pregnancy zone protein and alpha2-macroglobulin. J. Neurosci. Res. 2002, 70, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Vaglienti, M.V.; Subirada, P.V.; Barcelona, P.F.; Bonacci, G.; Sanchez, M.C. Quantification of Reactive Oxygen Species Using 2’,7’-Dichlorofluorescein Diacetate Probe and Flow-Cytometry in Müller Glial Cells. J. Vis. Exp. 2022, 13, 183. [Google Scholar] [CrossRef]
- Ridano, M.E.; Racca, A.C.; Flores-Martín, J.; Camolotto, S.A.; de Potas, G.M.; Genti-Raimondi, S.; Panzetta-Dutari, G.M. Chlorpyrifos modifies the expression of genes involved in human placental function. Reprod. Toxicol. 2012, 33, 331–338. [Google Scholar] [CrossRef]
- Subirada, P.V.; Vaglienti, M.V.; Joray, M.B.; Paz, M.C.; Barcelona, P.F.; Sánchez, M.C. Rapamycin and Resveratrol Modulate the Gliotic and Pro-Angiogenic Response in Müller Glial Cells Under Hypoxia. Front. Cell Dev. Biol. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Arnaoutova, I.; Kleinman, H.K. In vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010, 5, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Jyrkkänen, H.K.; Volger, O.L.; Leinonen, H.; Kivelä, A.M.; Häkkinen, S.K.; Woodcock, S.R.; Schopfer, F.J.; Horrevoets, A.J.; Ylä-Herttuala, S.; et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: Identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009, 284, 33233–33241. [Google Scholar] [CrossRef]
- Vazquez, M.M.; Gutierrez, M.v.; Salvatore, S.R.; Puiatti, M.; Dato, V.A.; Chiabrando, G.A.; Freeman, B.A.; Schopfer, F.J.; Bonacci, G. Nitro-oleic acid, a ligand of CD36, reduces cholesterol accumulation by modulating oxidized-LDL uptake and cholesterol efflux in RAW264.7 macrophages. Redox Biol. 2020, 36, 101591. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.C.; Holm, M.; Austeng, D.; Morken, T.S.; Zhou, T.E.; Beaudry-Richard, A.; Sierra, E.M.; Dammann, O.; Chemtob, S. Retinopathy of prematurity: Inflammation, choroidal degeneration, and novel promising therapeutic strategies. J. Neuroinflammation 2017, 14, 1–4. [Google Scholar] [CrossRef]
- Yamane, K.; Minamoto, A.; Yamashita, H.; Takamura, H.; Miyamoto-Myoken, Y.; Yoshizato, K.; Nabetani, T.; Tsugita, A.; Mishima, H.K. Proteome analysis of human vitreous proteins. Mol. Cell Proteomics. 2003, 2, 1177–1187. [Google Scholar] [CrossRef]
- Ridano, M.E.; Subirada, P.v.; Paz, M.C.; Lorenc, V.E.; Stupirski, J.C.; Gramajo, A.L.; Luna, J.D.; Croci, D.O.; Rabinovich, G.A.; Sánchez, M.C. Galectin-1 expression imprints a neurovascular phenotype in proliferative retinopathies and delineates responses to anti-VEGF. Oncotarget 2017, 8, 32505–32522. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res. 2015, 49, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Tah, V.; Orlans, H.O.; Hyer, J.; Casswell, E.; Din, N.; Sri Shanmuganathan, V.; Ramskold, L.; Pasu, S. Anti-VEGF Therapy and the Retina: An Update. J. Ophthalmol. 2015, 2015, 627674. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Aguilar, E.; Jacobson, R.; Trauger, S.A.; Friedlander, J.; Siuzdak, G.; Friedlander, M. Maintaining retinal astrocytes normalizes revascularization and prevents vascular pathology associated with oxygen-induced retinopathy. Glia 2010, 58, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, M.; Lemtalsi, T.; Toque, H.; Xu, Z.; Fulton, D.; Caldwell, R.; Caldwell, R. () NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Gross, S.; Chatterjee, A.; Wang, Y.; Lin, J.; Hammes, H.P. Transcription of Inflammatory Cytokine TNFα is Upregulated in Retinal Angiogenesis under Hyperoxia. Cell Physiol. Biochem. 2016, 39, 573–583. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Platt, D.H.; Matragoon, S.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Inhibition of NAD(P)H Oxidase Activity Blocks Vascular Endothelial Growth Factor Overexpression and Neovascularization during Ischemic Retinopathy. Am. J. Pathol. 2005, 167, 599–607. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.X.; Hartnett, M.E. Signaling pathways triggered by oxidative stress that mediate features of severe retinopathy of prematurity. JAMA Ophthalmol. 2013, 131, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Graziosi, A.; Perrotta, M.; Russo, D.; Gasparroni, G.; D’Egidio, C.; Marinelli, B.; di Marzio, G.; Falconio, G.; Mastropasqua, L.; Li Volti, G.; et al. Oxidative Stress Markers and the Retinopathy of Prematurity. J. Clin. Med. 2020, 9, 2711. [Google Scholar] [CrossRef]
- Ogihara, T.; Mino, M. Vitamin E and preterm infants. Free Radic. Biol. Med. 2022, 180, 13–32. [Google Scholar] [CrossRef]
- Khoo, N.K.H.; Li, L.; Salvatore, S.R.; Schopfer, F.J.; Freeman, B.A. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-κB signaling:A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 2295. [Google Scholar] [CrossRef]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Wright, M.M.; Kim, J.; Hock, T.D.; Leitinger, N.; Freeman, B.A.; Agarwal, A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009, 422, 353–361. [Google Scholar] [CrossRef]
- Tan, S.M.; Deliyanti, D.; Figgett, W.A.; Talia, D.M.; de Haan, J.B.; Wilkinson-Berka, J.L. Ebselen by modulating oxidative stress improves hypoxia-induced macroglial Müller cell and vascular injury in the retina. Exp. Eye Res. 2015, 136, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Fu, Z.J.; Lo, A.C.Y. Hypoxia-induced oxidative stress in ischemic retinopathy. Oxid. Med. Cell Longev. 2012, 2012, 426769. [Google Scholar] [CrossRef]
- Rudolph, T.K.; Rudolph, V.; Edreira, M.M.; Cole, M.P.; Bonacci, G.; Schopfer, F.J.; Woodcock, S.R.; Franek, A.; Pekarova, M.; Khoo, N.K.H.; et al. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.C.; Luna, J.D.; Barcelona, P.F.; Gramajo, A.L.; Juarez, P.C.; Riera, C.M.; Chiabrando, G.A. Effect of retinal laser photocoagulation on the activity of metalloproteinases and the alpha(2)-macroglobulin proteolytic state in the vitreous of eyes with proliferative diabetic retinopathy. Exp. Eye Res. 2007, 85, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Nedzvetskii, V.S.; Tuzcu, M.; Yasar, A.; Kirichenko, S.V. Increase of glial fibrillary acidic protein and S-100B in hippocampus and cortex of diabetic rats: Effects of vitamin E. Eur. J. Pharmacol. 2003, 462, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ho, G.; Zhang, Y.; Zhuo, L. In vivo imaging of retinal gliosis: A platform for diagnosis of PD and Screening of anti-PD compounds. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2010, 2010, 3049–3052. [Google Scholar]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.v. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS ONE 2010, 5, e9532. [Google Scholar] [CrossRef]
- Wang, T.; Tsirukis, D.I.; Sun, Y. Targeting Neuroinflammation in Neovascular Retinal Diseases. Front Pharmacol. 2020, 11, 234. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug. Discov. 2012, 11, 633–652. [Google Scholar]
- Nadeau-Vallée, M.; Quiniou, C.; Palacios, J.; Hou, X.; Erfani, A.; Madaan, A.; Sanchez, M.; Leimert, K.; Boudreault, A.; Duhamel, F.; et al. Novel Noncompetitive IL-1 Receptor-Biased Ligand Prevents Infection- and Inflammation-Induced Preterm Birth. J. Immunol. 2015, 195, 3402–3415. [Google Scholar] [CrossRef]
- Rivera, J.C.; Sitaras, N.; Noueihed, B.; Hamel, D.; Madaan, A.; Zhou, T.; Honoré, J.C.; Quiniou, C.; Joyal, J.S.; Hardy, P.; et al. Microglia and interleukin-1β in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Mantsounga, C.S.; Lee, C.; Neverson, J.; Sharma, S.; Healy, A.; Berus, J.M.; Parry, C.; Ceneri, N.M.; López-Giráldez, F.; Chun, H.J.; et al. Macrophage IL-1β promotes arteriogenesis by autocrine STAT3- and NF-κB-mediated transcription of pro-angiogenic VEGF-A. Cell Rep. 2022, 38, 110309. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kanai, H.; Sekiguchi, K.; Aihara, Y.; Yokoyama, T.; Arai, M.; Kanda, T.; Nagai, R.; Kurabayashi, M. Induction of VEGF gene transcription by IL-1 beta is mediated through stress-activated MAP kinases and Sp1 sites in cardiac myocytes. J Mol. Cell Cardiol. 2000, 32, 1955–1967. [Google Scholar] [CrossRef] [PubMed]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Gong, J.; Yoshida, T.; Eberhart, C.G.; Xu, Z.; Kombairaju, P.; Sporn, M.B.; Handa, J.T.; Duh, E.J. Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia-reperfusion injury. Free Radic. Biol. Med. 2011, 51, 216–224. [Google Scholar] [CrossRef]
- Wei, Y.; Gong, J.; Thimmulappa, R.K.; Kosmider, B.; Biswal, S.; Duh, E.J. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl. Acad. Sci. USA 2013, 110, E3910–E3918. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaglienti, M.V.; Subirada, P.V.; Joray, M.B.; Bonacci, G.; Sánchez, M.C. Protective Effect of NO2-OA on Oxidative Stress, Gliosis, and Pro-Angiogenic Response in Müller Glial Cells. Cells 2023, 12, 494. https://doi.org/10.3390/cells12030494
Vaglienti MV, Subirada PV, Joray MB, Bonacci G, Sánchez MC. Protective Effect of NO2-OA on Oxidative Stress, Gliosis, and Pro-Angiogenic Response in Müller Glial Cells. Cells. 2023; 12(3):494. https://doi.org/10.3390/cells12030494
Chicago/Turabian StyleVaglienti, María V., Paula V. Subirada, Mariana B. Joray, Gustavo Bonacci, and María C. Sánchez. 2023. "Protective Effect of NO2-OA on Oxidative Stress, Gliosis, and Pro-Angiogenic Response in Müller Glial Cells" Cells 12, no. 3: 494. https://doi.org/10.3390/cells12030494
APA StyleVaglienti, M. V., Subirada, P. V., Joray, M. B., Bonacci, G., & Sánchez, M. C. (2023). Protective Effect of NO2-OA on Oxidative Stress, Gliosis, and Pro-Angiogenic Response in Müller Glial Cells. Cells, 12(3), 494. https://doi.org/10.3390/cells12030494