
The Role of Growth Hormone and Insulin Growth Factor 1 in the Development of Non-Alcoholic Steato-Hepatitis: A Systematic Review

 , , and
, , and
Abstract
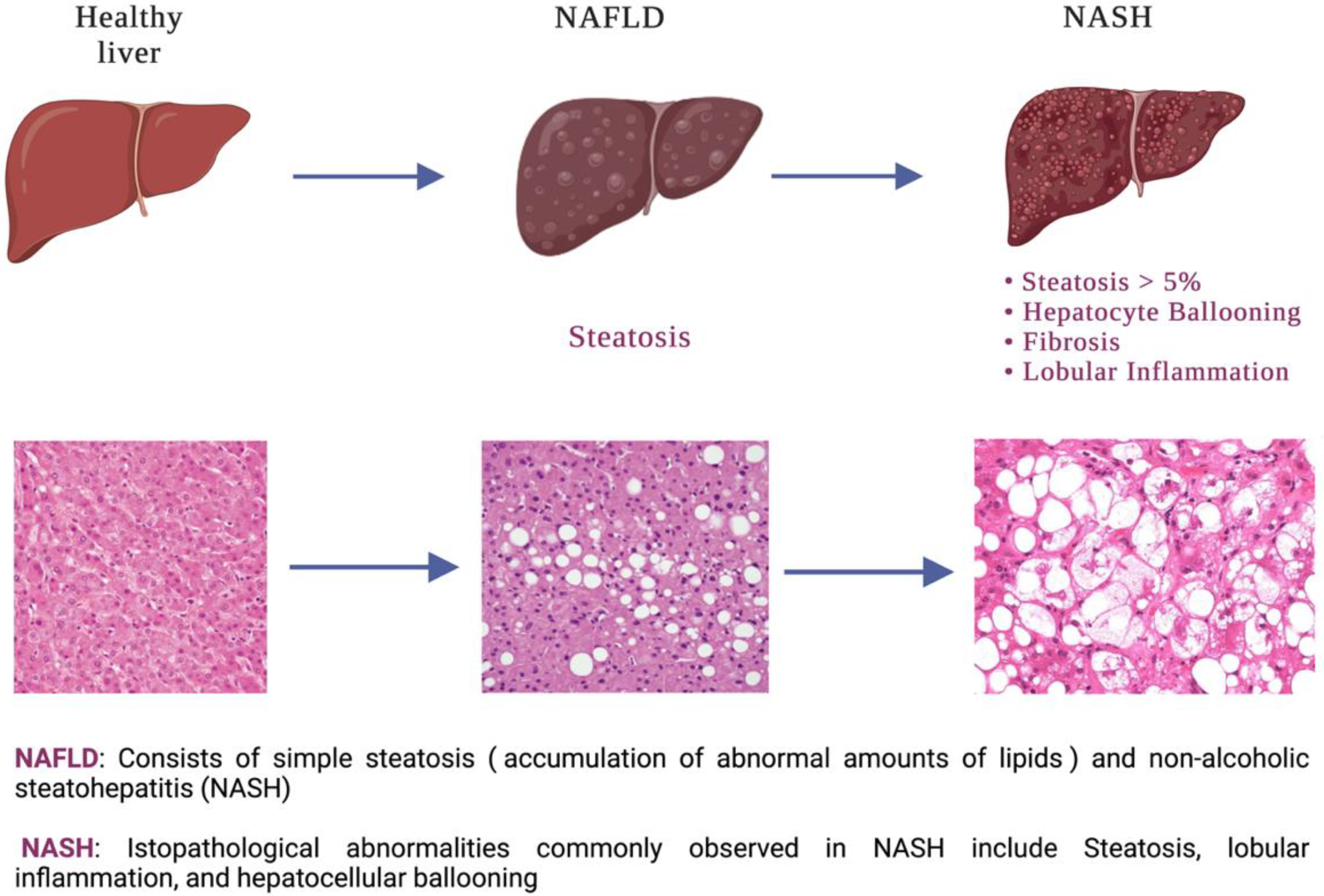
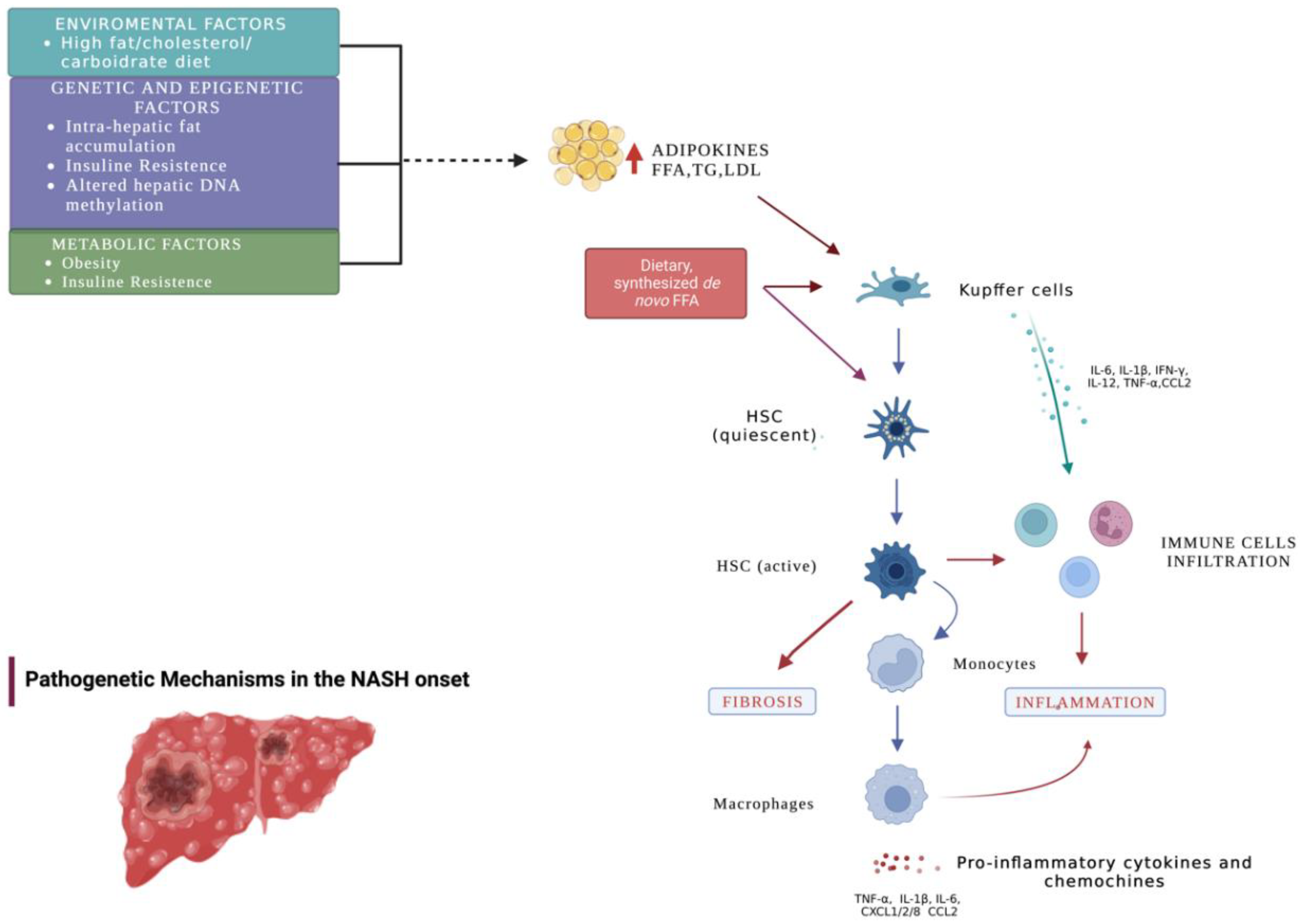
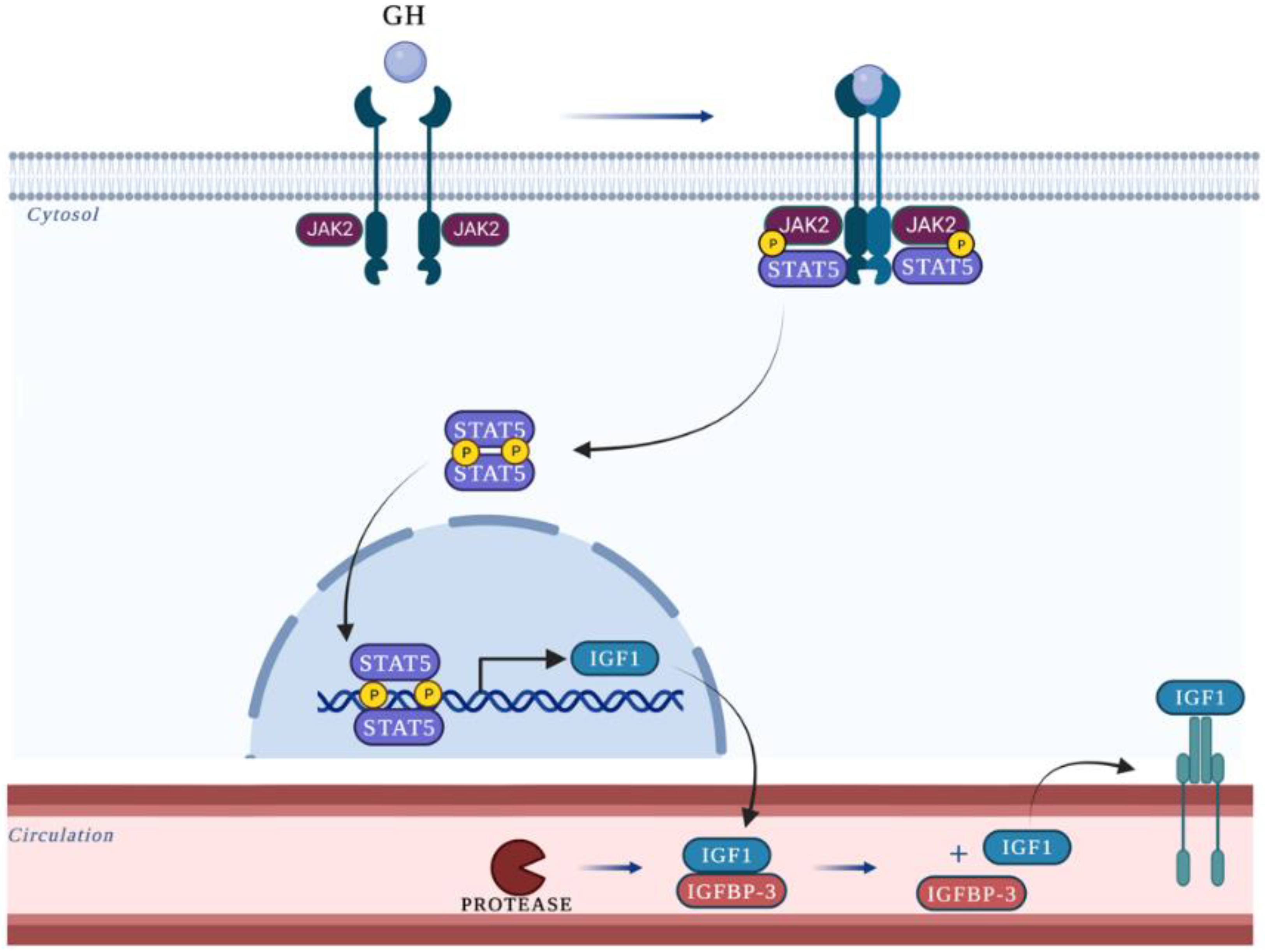
1. Introduction
2. Materials and Methods
2.1. Searches
2.2. Inclusion and Exclusion Criteria
2.3. Study Selection and Data Extraction
3. Results
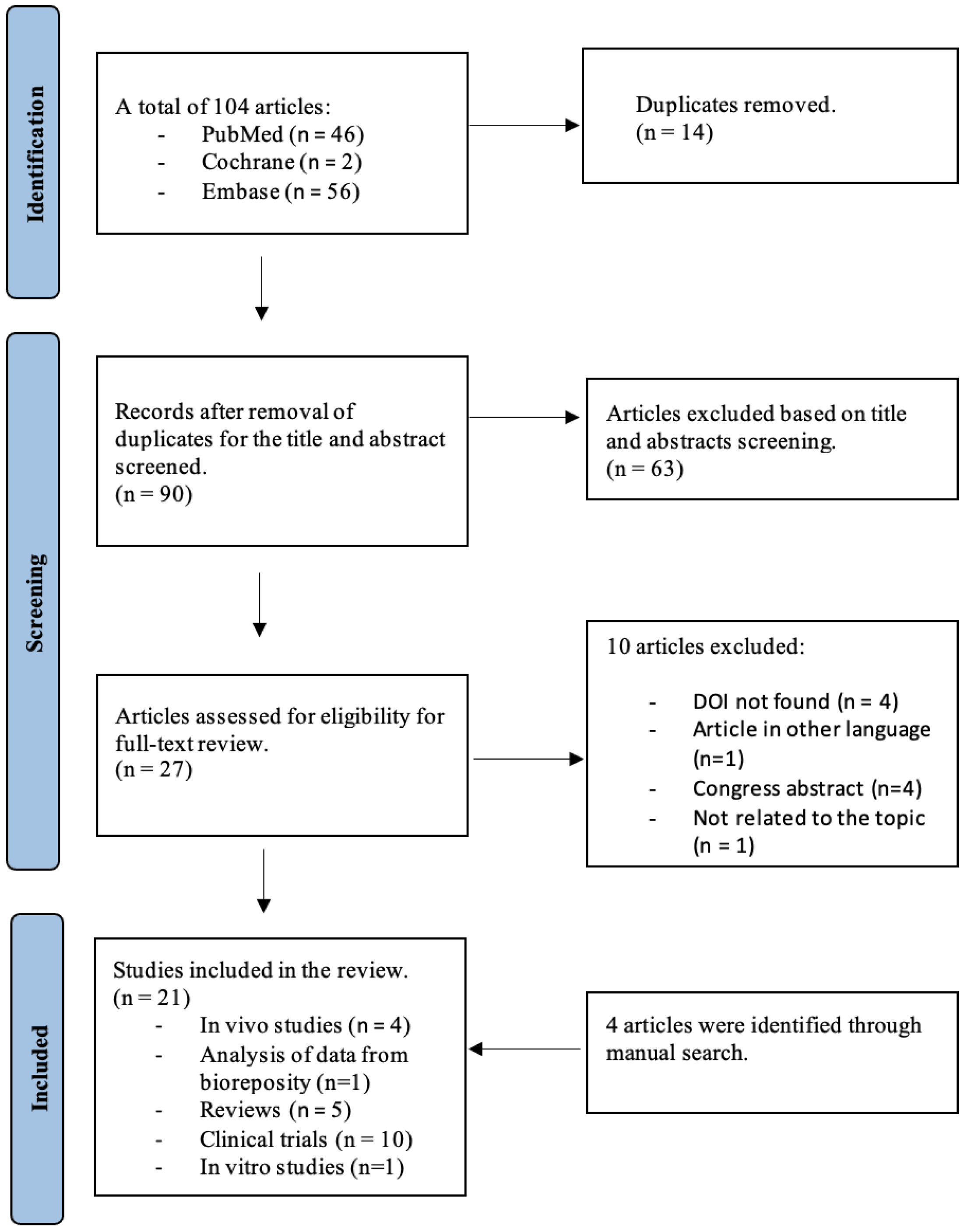
3.1. Searching Results
3.2. In Vitro and In Vivo Studies (Table 2)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study/Year | Techniques for Analysis of Results | Main Findings | p-Value |
|---|---|---|---|
| Nishizawa et al., 2012 [21] | Histological and biochemical analysis RT-PCR Immunochemical analysis | ↑ Liver triglyceride content (9 mg/g vs. 1mg/g) ↑ Serum AST and ALT, steatosis, and hepatic cell injury in GH-deficient rat (SDR) (mean ALT 30 IU/L vs. 12 IU/L; mean AST 250 IU/L vs. 100 IU/L) ↓ CPT-1, ↑ expression of enzymes for triglyceride synthesis ↑ Oxidative stress markers in SDR compared with those in the control GH and/or IGF-I administration improved all these changes | Liver triglyceride content: p < 0.001 Serum AST: p < 0.05 Serum ALT: p < 0.01 |
| Nishizawa et al., 2016 [26] | Histological analysis Immunoblotting pPCR | ↓ Tissue triglyceride content, ↓ cells showing ballooning necrosis, ↓ fibrosis in cells in mice NASH model treated by IGF-1(ballooning necrosis cell number/HPF reduced by circa 50%; tissue triglyceride reduced approximately by 20%) IGF-1-induced cellular senescence of HSCs IGF-1 treatment in the NASH mouse model induced a decrease in the expression of activated markers for HSCs (relative mRNA 𝛼SMA reduced by approximately 60%) p53 is necessary for the IGF-I-induced senescence in HSCs | Ballooning necrosis: cell number p < 0.03 Tissue triglyceride content p < 0.05 Expression of mRNA 𝛼SMA: p < 0.04 |
| Hosui et al., 2016 [25] | RT-PCR Western blot Immunohistochemistry and immunoblotting | ↑ CD36 in STAT5KO mice compared to those in control mice (more than 16-fold) In STAT5KO mice, CD36 gene expression is increased resulting in increased lipogenesis, fatty acid uptake, and steatosis. Improvement of abnormal lipid accumulation in STAT5/CD36 double KO mice compared to those in STAT5KO mice | |
| Fukunaga et al., 2018 [27] | RT-PCR Western blot Immunohistochemistry | IGF-1 increased ABCA1 mRNA expression 3 folds compared to those in the controls ABCA1 activity decreased in mice after administration of PolyEthylene Glycol (PEG), a GH receptor inhibitor. ↓ Cholesterol content in HepG2 cells treated by IGF-1 as compared to those in the control | ABCA expression p < 0.05 Cholesterol content: p < 0.05 |
| Sarmento-Cabral et al., 2021 [31] | Blood and hepatic lipid analysis Histological analysis | ↑ TG levels, hepatic fatty acids, and de novo lipogenesis in GHR knock-down mice compared with controls Improvement in whole-body lipid oxidation, fat mass, and insulin levels in male mice treated with IGF-1. ↑ALT levels, ↑steatosis, and hepatocyte ballooning (markers of liver injury) in GHR knock-down mice even after IGF-1 administration |
3.3. Human Studies (Table 3)
| Study/Year | Techniques for Analysis of Results | Main Findings | p-Value |
|---|---|---|---|
| Ichikawa et al., 2007 [17] | Clinical, laboratory and liver histology data | ↓ level of GH is associated with steatosis grade 2–3 (univariate RR = 0.196; multivariate RR = 0.199) ↓ level of IGF-1 is associated with fibrosis GH/IGF-1 ratio is significantly lower in patients with steatosis grade 2–3 compared to those with grade 1 No significant difference in GH/IGF-1 ratio among different grades of steatosis | Level of GH is associated with steatosis grade 2–3 univariate: p = 0.0269 multivariate; p = 0.0414 GH/IGF-1 ratio (p < 0.001) |
| García-Galiano et al., 2007 [18] | Clinical, biochemical, and histologic data | ↓ IGF-1 levels in patients with severe steatosis compared to those in healthy subjects and morbidly obese patients ↓ Concentration of IGF-1 in blood as compared to those in non-NASH and probable-NASH group Levels of IGF-1 <130 ng/mL as well as IGF-1 < 110 ng/mL are identified as independent predictors of hepatic steatosis and degree of NASH, respectively For IGF-1 < 130 ng/mL, the area below the ROC curve was found to be 0.75, while sensitivity and specificity were 0.68 and 0.79, respectively. For IGF-1 < 110 ng/mL, the area below the ROC curve was found to be 0.80, while sensitivity and specificity were 0.81 and 0.67, respectively. | IGF-1 levels (p = 0.024) Concentration of IGF (p = 0.006) |
| Koehler et al., 2012 [19] | Biochemical and histologic data | ↓ GH levels in NASH patients compared to those in controls (NASH with FS 0–1 patients had a median GH level of 0.10 ng/mL; NASH with FS ≥ 2 patients had a median GH level of 0.14 ng/mL; the controls group had a median GH level of 0.45 ng/mL) A normal GH level essentially excluded the presence of NASH with advanced fibrosis. | GH level in patients and control: p < 0.001 p < 0.001 |
| Nishizawa et al., 2012 [21] | Biochemical and histologic data | High prevalence of NAFLD among adult hypopituitary patients with GHD as compared to that in the control group (77% vs. 12%) ↓ Fibrotic marker concentrations, (p = 0.04) Improvement of histological changes in patients with NASH after GH replacement therapy) Six months after GH replacement therapy, serum liver enzyme concentrations were significantly decreased. | Prevalence of NAFLD: p < 0.001 Fibrotic marker concentrations: (p = 0.04) ALT: p < 0.001; AST: p < 0.005 and 𝛾-GTP: p < 0.05 |
| Cianfarani et al., 2014 [22] | Biochemical and histologic data | IGF-1 SDS levels were inversely related to the steatosis grade (r = −0.37), ballooning (r = −0.47), and NAS (r = −0.49). IGF-I/IGFBP-3 ratio was a significant predictor of liver inflammation (β = −0.285). ↓ levels of IGF-1 SDS and IGF-1/IGFBP-3 ratio in children with NASH and higher grades of steatosis | Steatosis grade: p < 0.002 Ballooning: p < 0.001 NAS: p < 0.001 IGF-I/IGFBP-3 ratio: p = 0.005 Levels of IGF-1 SDS: (p < 0.05) IGF-1/IGFBP-3 ratio: (p < 0.02) |
| Sumida et al., 2015 [24] | Clinical and histological data | Negative relationship between IGF-1 SDS and the activity of lobular inflammation (r = −0.134) and fibrosis (r = −0.362, p < 0.001); No relationship between the IGF-1:SDS and steatosis grade was detected | IGF-1 SDS: p < 0.001 Fibrosis: p < 0.001 |
| Dichtel et al., 2017 [11] | Biochemical and histologic data | ↓ Serum IGF-1 levels associated with lobular inflammation and hepatocyte ballooning (112 ± 47 ng/mL vs. 136 ± 57 ng/mL; 115 ± 48 ng/mL vs. 135 ± 57 ng/mL respectively) Subset analysis of patients presenting NASH demonstrated lower mean serum IGF-1 levels compared to the respective negative controls (115 ± 8 ng vs. 137 ± 8 ng) | Serum IGF-1 levels associated with lobular inflammation (p = 0.01), hepatocyte ballooning, (p = 0.05) Serum IGF-1 levels: p = 0.02 |
| Rufinatscha et al., 2018 [28] | Biochemical and histologic data | ↓ IGF-1 mRNA in patients with NASH when compared to patients with simple steatosis (approximative reduction of 66%). Among the 15 NASH patients, IGF-1 expression was characterized by an inverse relation to the grade of inflammation, but no statistical significance was found. GHR mRNA levels were comparable in patients with NASH and simple steatosis | IGF-1 mRNA: p < 0.05 IGF-1 expression: p = 0.25 |
| Polyzos et al., 2020 [29] | Biochemical and clinical measurements | ↓ IGF-1/intact IGFBP-3 ratio in NASH patients with fibrosis as compared to in controls with milder or no histologic lesions in the liver After performing a binary logistic regression, the IGF-1/intact IGFBP-3 ratio did not remain robustly associated with NASH or liver fibrosis (unadjusted and after adjusting for BMI and age) | IGF-1/intact IGFBP-3 ratio: p = 0.04 IGF-1/intact IGFBP-3 after performing a binary logistic regression (p = 0.06 unadjusted and p = 0.08 after adjusting for BMI and age) |
| Stanley et al., 2021 [30] | Analysis of data from a randomized clinical trial of GHRH. | ↓ Hepatic IGF-1 expression in individuals with higher grades of steatosis and higher NAS scores. ↓IGFBP2 and IGFBP4 (r = −0.49; r = −0.12) and ↑ IGFBP6 and IGFBP7 (r = 0.25; r = 0.35) with increasing steatosis Reduction of IGFBP2 after tesamorelin was associated with lower NAS score (r = 0.35) and hepatocellular ballooning grade (r = 0.37), but not with changes in lobular inflammation grade (r = 0.17) GHRH increased circulating IGFBP-1 and IGFBP-3 but decreased IGFBP-2 and IGFBP-6 | IGFBP2 and IGFBP4: p < 0.02, p < 0.02 IGFBP6 and IGFBP7: p < 0.005, p < 0.0001 IGFBP2 was associated with lower NAS score: (p = 0.02), hepatocellular ballooning grade (p = 0.02), but not with changes in lobular inflammation grade (p = 0.28) |
| Osganian et al., 2022 [33] | Gene expression analysis Immunohistochemistry | No difference in IGF-1 receptor or GH receptor gene expression across worsening stages of NAFLD/NASH compared to that in control ↓ IGF-1 gene expression across disease stages No difference in GH receptor staining intensity by the severity of NAFLD compared to that in the control group |
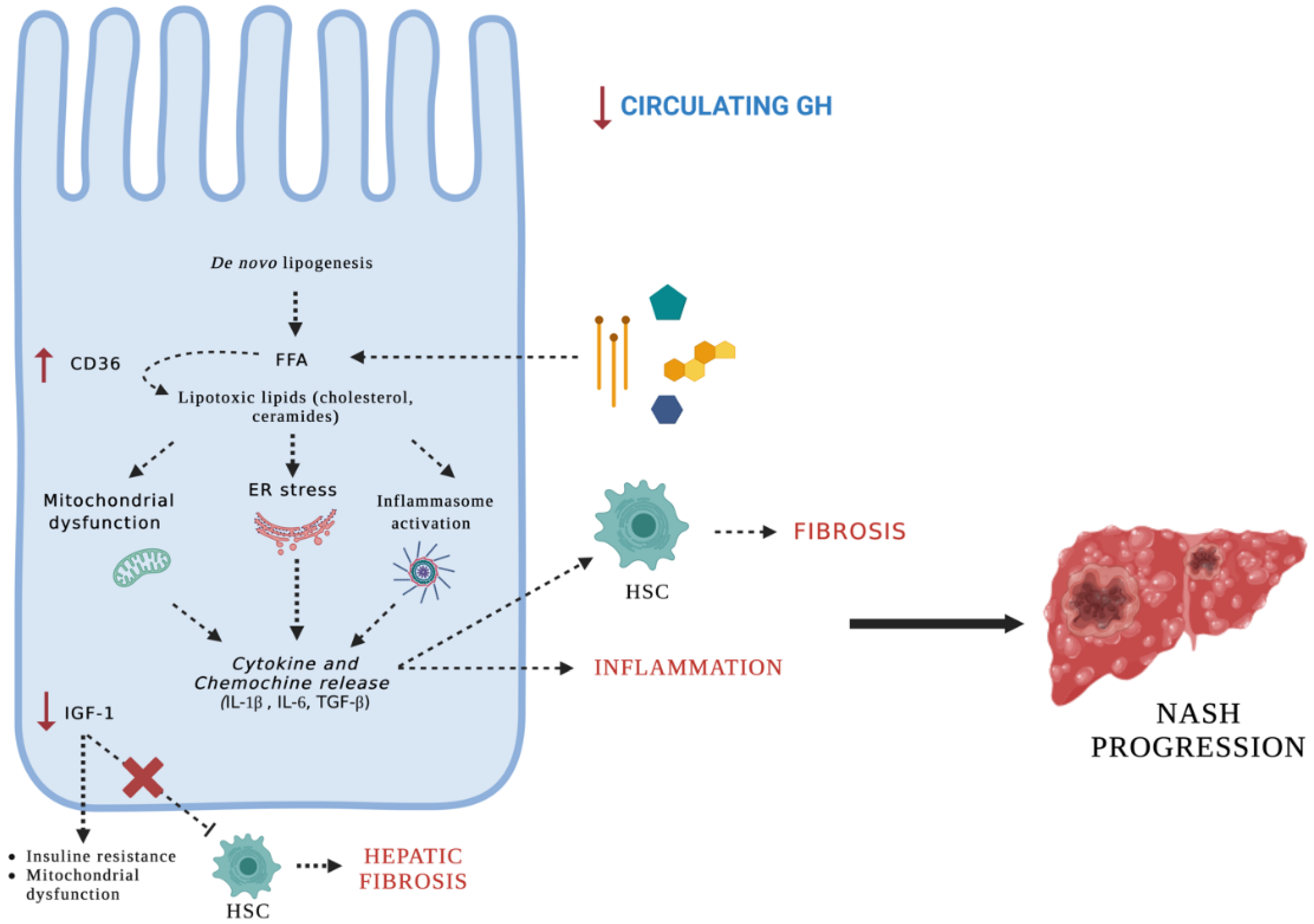
4. Discussion
Limitations and Strengths
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Takahashi, M.; Fukuoka, H.; Iguchi, G.; Kitazawa, R.; Takahashi, Y. GH-independent IGF-I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH-deficient rat model. Biochem. Biophys. Res. Commun. 2012, 423, 295–300. [Google Scholar] [CrossRef]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Smirne, C.; Croce, E.; Di Benedetto, D.; Cantaluppi, V.; Comi, C.; Sainaghi, P.P.; Minisini, R.; Grossini, E.; Pirisi, M. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. Livers 2022, 2, 30–76. [Google Scholar] [CrossRef]
- Wong, R.J.; Singal, A.K. Trends in Liver Disease Etiology Among Adults Awaiting Liver Transplantation in the United States, 2014–2019. JAMA Netw. Open 2020, 3, e1920294. [Google Scholar] [CrossRef]
- Mallet, V.; Parlati, L.; Martinino, A.; Scarano Pereira, J.P.; Jimenez, C.N.; Sakka, M.; Bouam, S.; Retbi, A.; Krasteva, D.; Meritet, J.-F.; et al. Burden of liver disease progression in hospitalized patients with type 2 diabetes mellitus. J. Hepatol. 2022, 76, 265–274. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Dichtel, L.E.; Corey, K.E.; Misdraji, J.; Bredella, M.A.; Schorr, M.; Osganian, S.A.; Young, B.J.; Sung, J.C.; Miller, K.K. The Association Between IGF-1 Levels and the Histologic Severity of Nonalcoholic Fatty Liver Disease. Clin. Transl. Gastroenterol. 2017, 8, e217. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Hao, J.-W.; Wang, J.; Guo, H.; Zhao, Y.-Y.; Sun, H.-H.; Li, Y.-F.; Lai, X.-Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakao, K.; Hamasaki, K.; Furukawa, R.; Tsuruta, S.; Ueda, Y.; Taura, N.; Shibata, H.; Fujimoto, M.; Toriyama, K.; et al. Role of growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease. Hepatol. Int. 2007, 1, 287–294. [Google Scholar] [CrossRef]
- García-Galiano, D.; Sánchez-Garrido, M.A.; Espejo, I.; Montero, J.L.; Costán, G.; Marchal, T.; Membrives, A.; Gallardo-Valverde, J.M.; Muñoz-Castañeda, J.R.; Arévalo, E.; et al. IL-6 and IGF-1 are independent prognostic factors of liver steatosis and non-alcoholic steatohepatitis in morbidly obese patients. Obes. Surg. 2007, 17, 493–503. [Google Scholar] [CrossRef]
- Koehler, E.; Swain, J.; Sanderson, S.; Krishnan, A.; Watt, K.; Charlton, M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: An endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012, 32, 279–286. [Google Scholar] [CrossRef]
- Takahashi, Y. Essential roles of growth hormone (GH) and insulin-like growth factor-I (IGF-I) in the liver. Endocr. J. 2012, 59, 955–962. [Google Scholar] [CrossRef]
- Nishizawa, H.; Iguchi, G.; Murawaki, A.; Fukuoka, H.; Hayashi, Y.; Kaji, H.; Yamamoto, M.; Suda, K.; Takahashi, M.; Seo, Y.; et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur. J. Endocrinol. 2012, 167, 67–74. [Google Scholar] [CrossRef]
- Cianfarani, S.; Inzaghi, E.; Alisi, A.; Germani, D.; Puglianiello, A.; Nobili, V. Insulin-Like Growth Factor-I and -II Levels Are Associated with the Progression of Nonalcoholic Fatty Liver Disease in Obese Children. J. Pediatr. 2014, 165, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Xanthakos, S.A.; Crimmins, N.A.; Chernausek, S.D. Abnormalities in the growth hormone axis and risk of nonalcoholic steatohepatitis: Active player or innocent bystander? J. Pediatr. 2014, 165, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yonei, Y.; Tanaka, S.; Mori, K.; Kanemasa, K.; Imai, S.; Taketani, H.; Hara, T.; Seko, Y.; Ishiba, H.; et al. Lower levels of insulin-like growth factor-1 standard deviation score are associated with histological severity of non-alcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 771–781. [Google Scholar] [CrossRef]
- Hosui, A.; Tatsumi, T.; Hikita, H.; Saito, Y.; Hiramatsu, N.; Tsujii, M.; Hennighausen, L.; Takehara, T. STAT5 plays a crucial role in hepatic lipid metabolism through regulation of CD36 expression: STAT5 negatively regulates CD36. Hepatol. Res. 2016, 47, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605. [Google Scholar] [CrossRef]
- Fukunaga, K.; Imachi, H.; Lyu, J.; Dong, T.; Sato, S.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Yonezaki, K.; Matsunaga, T.; et al. IGF1 suppresses cholesterol accumulation in the liver of growth hormone-deficient mice via the activation of ABCA1. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E1232–E1241. [Google Scholar] [CrossRef] [PubMed]
- Rufinatscha, K.; Ress, C.; Folie, S.; Haas, S.; Salzmann, K.; Moser, P.; Dobner, J.; Weiss, G.; Iruzubieta, P.; Arias-Loste, M.T.; et al. Metabolic effects of reduced growth hormone action in fatty liver disease. Hepatol. Int. 2018, 12, 474–481. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Perakakis, N.; Boutari, C.; Kountouras, J.; Ghaly, W.; Anastasilakis, A.D.; Karagiannis, A.; Mantzoros, C.S. Targeted Analysis of Three Hormonal Systems Identifies Molecules Associated with the Presence and Severity of NAFLD. J. Clin. Endocrinol. Metab. 2020, 105, e390–e400. [Google Scholar] [CrossRef]
- Stanley, T.L.; Fourman, L.T.; Zheng, I.; McClure, C.M.; Feldpausch, M.N.; Torriani, M.; Corey, K.E.; Chung, R.T.; Lee, H.; Kleiner, D.E.; et al. Relationship of IGF-1 and IGF-Binding Proteins to Disease Severity and Glycemia in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2021, 106, e520–e533. [Google Scholar] [CrossRef]
- Sarmento-Cabral, A.; Del Rio-Moreno, M.; Vazquez-Borrego, M.C.; Mahmood, M.; Gutierrez-Casado, E.; Pelke, N.; Guzman, G.; Subbaiah, P.V.; Cordoba-Chacon, J.; Yakar, S.; et al. GH directly inhibits steatosis and liver injury in a sex-dependent and IGF1-independent manner. J. Endocrinol. 2021, 248, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Dichtel, L.E.; Cordoba-Chacon, J.; Kineman, R.D. Growth Hormone and Insulin-Like Growth Factor 1 Regulation of Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2022, 107, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Osganian, S.A.; Subudhi, S.; Masia, R.; Drescher, H.K.; Bartsch, L.M.; Chicote, M.L.; Chung, R.T.; Gee, D.W.; Witkowski, E.R.; Bredella, M.A.; et al. Expression of IGF-1 receptor and GH receptor in hepatic tissue of patients with nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Growth Horm. IGF Res. 2022, 65, 101482. [Google Scholar] [CrossRef]
- Doycheva, I.; Erickson, D.; Watt, K.D. Growth hormone deficiency and NAFLD: An overlooked and underrecognized link. Hepatol. Commun. 2022, 6, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone–STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. N. Y. Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- Rotwein, P. Mapping the growth hormone--Stat5b--IGF-I transcriptional circuit. Trends Endocrinol. Metab. TEM 2012, 23, 186–193. [Google Scholar] [CrossRef]
- Feigerlova, E.; Hwa, V.; Derr, M.A.; Rosenfeld, R.G. Current issues on molecular diagnosis of GH signaling defects. Endocr. Dev. 2013, 24, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [CrossRef]
- Juul, A.; Scheike, T.; Davidsen, M.; Gyllenborg, J.; Jørgensen, T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: A population-based case-control study. Circulation 2002, 106, 939–944. [Google Scholar] [CrossRef]
- Kucera, R.; Topolcan, O.; Pecen, L.; Kinkorova, J.; Svobodova, S.; Windrichova, J.; Fuchsova, R. Reference values of IGF1, IGFBP3 and IGF1/IGFBP3 ratio in adult population in the Czech Republic. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 444, 271–277. [Google Scholar] [CrossRef]
- Zeng, M.-D.; Lu, L.-G.; Mao, Y.-M.; Qiu, D.-K.; Li, J.-Q.; Wan, M.-B.; Chen, C.-W.; Wang, J.-Y.; Cai, X.; Gao, C.-F.; et al. Prediction of significant fibrosis in HBeAg-positive patients with chronic hepatitis B by a noninvasive model. Hepatology 2005, 42, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Savastano, S.; Di Somma, C.; Pizza, G.; De Rosa, A.; Nedi, V.; Rossi, A.; Orio, F.; Lombardi, G.; Colao, A.; Tarantino, G. Liver-spleen axis, insulin-like growth factor-(IGF)-I axis and fat mass in overweight/obese females. J. Transl. Med. 2011, 9, 136. [Google Scholar] [CrossRef]
- Marques, V.; Afonso, M.B.; Bierig, N.; Duarte-Ramos, F.; Santos-Laso, Á.; Jimenez-Agüero, R.; Eizaguirre, E.; Bujanda, L.; Pareja, M.J.; Luís, R.; et al. Adiponectin, Leptin, and IGF-1 Are Useful Diagnostic and Stratification Biomarkers of NAFLD. Front. Med. 2021, 8, 683250. [Google Scholar] [CrossRef]
- Thissen, J.P.; Verniers, J. Inhibition by interleukin-1 beta and tumor necrosis factor-alpha of the insulin-like growth factor I messenger ribonucleic acid response to growth hormone in rat hepatocyte primary culture. Endocrinology 1997, 138, 1078–1084. [Google Scholar] [CrossRef]
- Anwar, A.; Zahid, A.A.; Scheidegger, K.J.; Brink, M.; Delafontaine, P. Tumor necrosis factor-alpha regulates insulin-like growth factor-1 and insulin-like growth factor binding protein-3 expression in vascular smooth muscle. Circulation 2002, 105, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Lelbach, A.; Scharf, J.G.; Ramadori, G. Regulation of insulin-like growth factor-I and of insulin-like growth factor binding protein-1, -3 and -4 in cocultures of rat hepatocytes and Kupffer cells by interleukin-6. J. Hepatol. 2001, 35, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Liu, J.L.; Fernandez, A.M.; Wu, Y.; Schally, A.V.; Frystyk, J.; Chernausek, S.D.; Mejia, W.; Le Roith, D. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes 2001, 50, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Fahlbusch, P.; Knebel, B.; Hörbelt, T.; Barbosa, D.M.; Nikolic, A.; Jacob, S.; Al-Hasani, H.; Van de Velde, F.; Van Nieuwenhove, Y.; Müller-Wieland, D.; et al. Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. Int. J. Mol. Sci. 2020, 21, 4144. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef]
- Drivdahl, R.; Haugk, K.H.; Sprenger, C.C.; Nelson, P.S.; Tennant, M.K.; Plymate, S.R. Suppression of growth and tumorigenicity in the prostate tumor cell line M12 by overexpression of the transcription factor SOX9. Oncogene 2004, 23, 4584–4593. [Google Scholar] [CrossRef]
- Ruan, W.; Xu, E.; Xu, F.; Ma, Y.; Deng, H.; Huang, Q.; Lv, B.; Hu, H.; Lin, J.; Cui, J.; et al. IGFBP7 plays a potential tumor suppressor role in colorectal carcinogenesis. Cancer Biol. Ther. 2007, 6, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Liso, A.; Capitanio, N.; Gerli, R.; Conese, M. From fever to immunity: A new role for IGFBP-6? J. Cell. Mol. Med. 2018, 22, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Shen, F.; Weinfeld, M.; Sergi, C. Insulin Growth Factor Binding Protein 7 (IGFBP7)-Related Cancer and IGFBP3 and IGFBP7 Crosstalk. Front. Oncol. 2020, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E. Vitamin E treatment of nonalcoholic steatohepatitis in children: A pilot study. J. Pediatr. 2000, 136, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-analysis: Pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2012, 35, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Kraenzlin, M.E.; Bodmer, M.; Jick, S.S.; Jick, H.; Meier, C.R. Use of thiazolidinediones and fracture risk. Arch. Intern. Med. 2008, 168, 820–825. [Google Scholar] [CrossRef]
- Lebovitz, H.E.; Dole, J.F.; Patwardhan, R.; Rappaport, E.B.; Freed, M.I. Rosiglitazone Clinical Trials Study Group Rosiglitazone monotherapy is effective in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Loke, Y.K.; Furberg, C.D. Thiazolidinediones and heart failure: A teleo-analysis. Diabetes Care 2007, 30, 2148–2153. [Google Scholar] [CrossRef]
- Xue, J.; Liang, S.; Ma, J.; Xiao, Y. Effect of growth hormone therapy on liver enzyme and other cardiometabolic risk factors in boys with obesity and nonalcoholic fatty liver disease. BMC Endocr. Disord. 2022, 22, 49. [Google Scholar] [CrossRef]
- Adams, L.A.; Angulo, P. Recent concepts in non-alcoholic fatty liver disease. Diabet. Med. 2005, 22, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]





| Study/Year | Study Design | Country | N° of Patients with NAFLD/NASH | N° of Controls | M/F Ratio | Mean Population Age (±SD) | Research Question | Method of Diagnosis | Comment |
|---|---|---|---|---|---|---|---|---|---|
| Ichikawa et al., 2007 [17] | CS | Japan | 55 patients with NAFLD | - | 20 males 35 females | Fibrosis stage 0–1: 47.5 ± 16.7 Fibrosis stage 2–3: 55.4 ± 17.5 | Role of GH, IGF-1, IGFBP-3 in NAFLD development | Percutaneous liver biopsy | |
| García-Galiano et al., 2007 [18] | CS | Spain | 36 morbidly obese patients (13 with probable NASH and 9 with NASH) | 12 healthy subjects | - | - | Association between the serological levels of TNF-α, IL-6, and IGF-1 with steatosis and NASH | Liver biopsy during surgery | |
| Koehler et al., 2012 [19] | CS | USA | 160 patients scheduled for bariatric surgery (72 with NASH and 72 with simple steatosis) | - | 24 males 136 females | Normal histology: 50 ± 13.2 S.S.: 47.8 ± 11.1 NASH and FS 0-1: 46.4 ± 10 NASH and FS >2: 50.7 ± 10.7 | Potential endocrine basis of steatohepatitis with advanced fibrosis in NAFLD | Liver biopsy | |
| Nishizawa et al., 2012 [4] | VIVO | Japan | - | - | - | - | Effect of GH and IGF-1 administration on the liver of GH-deficient rats | 5 GH-deficient rats (S.D.R.) and 5 age-matched rats as control. | |
| Takahashi, 2012 [20] | R | Japan | - | - | - | - | Role of GH and IGF-1 in the liver | - | |
| Nishizawa et al., 2012 [21] | CS | Japan | 66 patients with GHD. | 1994 healthy subjects | 32 males 34 females | Normal histology: 44.8 ± 16.0 NAFLD: 48.1 ± 18.0 NASH: 44.6 ± 6.4 | Prevalence of NAFLD/NASH in adult hypopituitary patients with GHD | Ultrasonography and liver biopsy | |
| Cianfarani et al., 2014 [22] | CS | Italy | 99 obese children (14 with NASH) | - | 57 males 42 females | 8.73 ± 1.98 | Correlate circulating levels of IGF-1, IGF2, and IGFBP3 with NASH | Liver biopsy | |
| Xanthakos et al., 2014 [23] | R | USA | - | - | - | - | Correlation between abnormalities in GH axis and NASH | - | |
| Sumida et al., 2015 [24] | CS | Japan | 199 Japanese patients with NAFLD | 2911 healthy people | 92 males 107 females | NAFLD: 59 ± 10 | Correlation between levels of IGF-1 SDS and histological severity of NAFLD | Liver biopsy | |
| Hosui et al., 2016 [25] | IVIV | Japan | - | - | - | - | Role of STAT5 in hepatic lipid metabolism | - | STAT5KO mice and their littermates as controls. |
| Nishizawa et al., 2016 [26] | IVIV | Japan | - | - | - | - | Effect of IGF-1 on NASH and cirrhotic models and the underlying mechanisms | - | NASH model, methionine-choline-deficient diet-fed db/db mice, and cirrhotic model, dimethylnitrosamine-treated mice |
| Takahashi, 2017 [2] | R | Japan | - | - | - | - | Role of GH and IGF-1 in the liver | - | |
| Dichtel et al., 2017 [11] | CS | USA | 121 patients (80 with NASH and 41 with simple steatosis) | 21 subjects | 66 males 76 postmenopausal females | Normal histology: 50 ± 10 S.S.: 55 ± 8 NASH: 50 ± 11 | Clarify the relationship between the histological severity of NAFLD and serum IGF-1 levels | Liver biopsy | |
| Fukunaga et al., 2018 [27] | IVIV | Japan | - | - | - | - | Determine the effects of IGF-1 on ABCA1 expression in GH-deficient mice | - | 3 groups (n = 5 each) of 8-week-old mice: (1) control with high-fat diet (H.F.D.) (2) H.F.D. + P.E.G. (3) H.F.D. + P.E.G. + IGF-1 |
| Rufinatscha et al., 2018 [28] | CS | Austria | 29 obese patients (15 with NASH and 14 with simple steatosis) | - | Female population exclusively | S.S.: 37.8 ± 11.8 NASH: 45.1 ± 8.9 | Role of hepatic GH signaling and its metabolic consequences in patients with NAFLD | Liver biopsy | |
| Polyzos et al., 2020 [29] | CS | Greece | 31 patients (16 with NASH and 15 with simple steatosis) | 50 subjects (24 lean controls and 26 obese controls) | 19 males 62 females | Normal histology: 56.6 ± 12.5 NASH: 56.6 ± 7.1 | Evaluate hormones levels in histologically confirmed NASH patients versus S.S. patients versus controls | Liver biopsy | |
| Stanley TL et al., 2021 [30] | RCT | USA | 61 subjects with HIV and hepatic steatosis | - | Predominantly male | 53 ± 7 | Clarify the relationships between hepatic expression of IGF-1 and IGFBPs and evaluate the effect of GHRH therapy in adults with NAFLD | Liver biopsy | |
| Sarmento-Cabral et al., 2021 [31] | VIVO | USA | - | - | - | - | Test the IGF-1-independent role of hepatocyte GHR signaling | - | Mice with adult-onset, hepatocyte-specific GHR knock-down (aHepGHRkd) treated with a vector expressing rat IGF-1 targeted specifically to hepatocytes |
| Dichtel et al., 2022 [32] | R | USA | - | - | - | - | Roles of GH and IGF-1 in the liver and their potential application for the treatment of NASH | - | |
| Osganian et al., 2022 [33] | * | USA | 318 patients for the gene expression cohort and 30 for the immunohistochemistry cohort | - | - | Normal histology: 41.6 ± 11.5 S.S.: 45.2 ± 11.7 NASH F0: 43.9 ± 12.2 NASH F1-F4: 45.1 ± 12.9 | IGF-1 receptor and GH receptor physiology in patients with NAFLD and NASH | Liver biopsy | |
| Doycheva et al., 2022 [34] | R | USA | - | - | - | - | Pathophysiologic mechanisms of GH in NAFLD, NAFLD association with AGHD, and effect of GH treatment in patients with NAFLD | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cristin, L.; Montini, A.; Martinino, A.; Scarano Pereira, J.P.; Giovinazzo, F.; Agnes, S. The Role of Growth Hormone and Insulin Growth Factor 1 in the Development of Non-Alcoholic Steato-Hepatitis: A Systematic Review. Cells 2023, 12, 517. https://doi.org/10.3390/cells12040517
Cristin L, Montini A, Martinino A, Scarano Pereira JP, Giovinazzo F, Agnes S. The Role of Growth Hormone and Insulin Growth Factor 1 in the Development of Non-Alcoholic Steato-Hepatitis: A Systematic Review. Cells. 2023; 12(4):517. https://doi.org/10.3390/cells12040517
Chicago/Turabian StyleCristin, Luca, Amalia Montini, Alessandro Martinino, Juan Pablo Scarano Pereira, Francesco Giovinazzo, and Salvatore Agnes. 2023. "The Role of Growth Hormone and Insulin Growth Factor 1 in the Development of Non-Alcoholic Steato-Hepatitis: A Systematic Review" Cells 12, no. 4: 517. https://doi.org/10.3390/cells12040517
APA StyleCristin, L., Montini, A., Martinino, A., Scarano Pereira, J. P., Giovinazzo, F., & Agnes, S. (2023). The Role of Growth Hormone and Insulin Growth Factor 1 in the Development of Non-Alcoholic Steato-Hepatitis: A Systematic Review. Cells, 12(4), 517. https://doi.org/10.3390/cells12040517