

Transition from Animal-Based to Human Induced Pluripotent Stem Cells (iPSCs)-Based Models of Neurodevelopmental Disorders: Opportunities and Challenges



Abstract
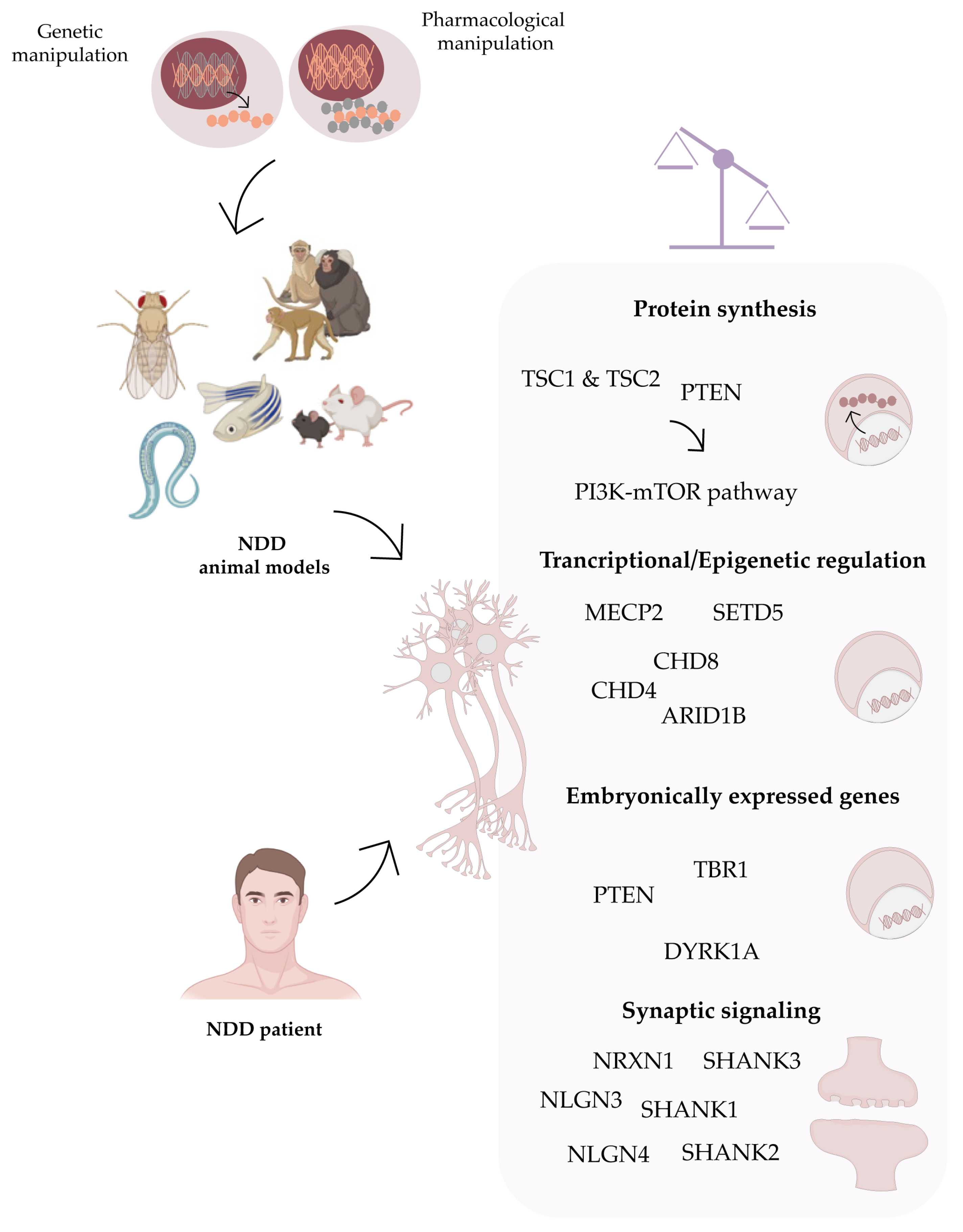
:1. Neurodevelopmental Disorders (NDDs)
2. Modeling NDDs with Human Induced Pluripotent Stem Cells (hIPSCs)
2.1. Human Induced Pluripotent Stem Cells (hIPSCs)
2.2. Current Methodological Approaches to Differentiate Neuronal Cells from hIPSCs—An Overview
2.2.1. Source Cells—Selection
2.2.2. Somatic Cell Reprogramming into hIPSCs

{kind=link}
{kind=link}
{kind=link}
| Reprogramming Method | Delivery | Type | Efficiency | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Retro- and lentivirus | Integrative transfection | Viral | Stable transgene expression [113] | Integration into the genome | |
| Increased risk of mutagenesis and tumorigenesis [113] | |||||
| Adenovirus | Non-integrative transfection | No integration | |||
| Well-defined biology | Not all cell types respond equally | ||||
| Genetic stability | Possibility of genome integration | ||||
| Easy large-scale production | Transient expression due to rapid clearance from dividing cells [129,130,131,132,133] | ||||
| Safe to use in clinical trials [134] | |||||
| AAV | Absence of immune/toxic reactions | Require a helper virus to replicate | |||
| Decreased titer production | |||||
| Stable transgene expression | Limited packaging capacity | ||||
| Safe to use in clinical trials [135,136] | Possibility of genome integration [134,136,137] | ||||
| SeV | No integration | ||||
| Non-pathogenic to humans | Difficult to remove from the generated hIPSCs | ||||
| Cytoplasmic replicative cycle | Cytotoxicity | ||||
| Low propensity for genomic/epigenetic aberrations | Difficult to work with | ||||
| High and fast protein expression | Enhanced fusogenicity and immunogenicity | ||||
| High transduction efficiency | Sensitivity to transgenic sequences [138,139] | ||||
| Fast cellular uptake | Low capacity to cross the cell membrane Lack of nuclear localization Challenging production of pure proteins Poor solubility and stability Sequestration of the transduced reprogramming proteins [117,118,140,141,142] | ||||
| Ideal transgene expression [116,138,143,144,145,146,147] | |||||
| Recombinant proteins | Protein | Low capacity to cross the cell membrane | |||
| Lack of nuclear localization | |||||
| No Integration | Challenging production of pure proteins | ||||
| Virus free [118] | Poor solubility and stability | ||||
| Sequestration of the transduced reprogramming proteins [117,118,140,141,142] | |||||
| mRNA transfections | mRNA | No integration | Difficult to work with | ||
| Virus free | |||||
| Low reprogramming time | Triggers immune system response | ||||
| Safe and high transduction [119,148,149,150] | Need for feeder cells and animal-derived molecules [151,152,153,154,155,156,157,158,159,160] | ||||
| Plasmid transfection | DNA | Transient expression of reprogramming factors | |||
| Virus free | Variation of transfection efficiency between cells | ||||
| No vulnerability to exonucleases [161] | Large size | ||||
| Lack of self-replication requiring multiple transfections [161,162] | |||||
| Mini-circle vectors | No integration | ||||
| Virus free | |||||
| High transgene expression | Lack of self-replication capacity | ||||
| Easy to synthetize and deliver | Decreased expression time | ||||
| Small size | Require multiple transfections [113,163,164,165] | ||||
| Less prone to transcriptional silencing | |||||
| Controlled concentration and application time [120,166,167] | |||||
| Episomal vectors | No integration | Low efficiency [121,137] | |||
| Virus free | |||||
| Single transfection | |||||
| Long-term, stable transgene expression | |||||
| Fast protein expression | |||||
| Absence of genome manipulation | |||||
| Lack of regulatory constraints in the target gene [121,137] | |||||
| Transposons | Stable integration | Possible reintroduction in the genome [168] | |||
| Virus free | |||||
| Carry large cargoes | |||||
| Single transfection with long-term, strong gene expression | |||||
| Inexpensive | |||||
| Easy to work with | |||||
| Removal of transgene cassette without induction of genetic mutations | |||||
| Low immunogenicity [137,169,170] | |||||
| Liposomal magnetofection | No integration | Low efficiency [171] | |||
| Virus free | |||||
| Single transfection with low immunogenicity [171,172] |
2.2.3. hIPSCs Differentiation into Neuronal Populations
2.2.4. Experimental Design
2.2.5. Gene-Editing Techniques Applicable to hIPSCs
2.2.6. Quality vs. Quantity, Cost-Associated, Time Needed
2.2.7. Genetic/Epigenetic Instability
2.2.8. hIPSC-Derived 3D Brain Organoids
2.3. hIPSC Models for the Study of NDDs
2.3.1. hIPSC Models of Autism Spectrum Disorders (ASD)
2.3.2. hIPSC Models of Rett Syndrome (RTT)
2.3.3. hIPSC Models of Down Syndrome
2.4. Tackling Molecular Pathways Affected in NDDs Using hIPSCs
2.4.1. High-Throughput Screenings Using hIPSCs (HTS)
2.4.2. Molecular Pathways Affected in NDDs—Insight from hIPSC-Based Studies
Autism Spectrum Disorders (ASD)
Rett Syndrome
Down Syndrome
2.4.3. Other NDDs
2.4.4. Translation to Clinics
2.5. Drug Screenings for NDDs Using hIPSCs
3. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Mullin, A.P.; Gokhale, A.; Moreno-De-Luca, A.; Sanyal, S.; Waddington, J.L.; Faundez, V. Neurodevelopmental disorders: Mechanisms and boundary definitions from genomes, interactomes and proteomes. Transl. Psychiatry 2013, 3, e329. [Google Scholar] [CrossRef] [PubMed]
- Francés, L.; Quintero, J.; Fernández, A.; Ruiz, A.; Caules, J.; Fillon, G.; Hervás, A.; Soler, C.V. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: A systematic review in accordance with the PRISMA criteria. Child Adolesc. Psychiatry Ment. Health 2022, 16, 1–15. [Google Scholar] [CrossRef]
- Zablotsky, B.; Black, L.I.; Maenner, M.J.; Schieve, L.A.; Danielson, M.L.; Bitsko, R.H.; Blumberg, S.J.; Kogan, M.D.; Boyle, C.A. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics 2019, 144, e20190811. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, X.; Wu, H.; Xun, G.; Ou, J.; Zhang, Q.; Li, H.; Bai, T.; Hu, Z.; Zou, X.; et al. Genotype and phenotype correlations for SHANK3 de novo mutations in neurodevelopmental disorders. Am. J. Med. Genet. Part A 2018, 176, 2668–2676. [Google Scholar] [CrossRef]
- Casanova, E.L.; Gerstner, Z.; Sharp, J.L.; Casanova, M.F.; Feltus, F.A. Widespread Genotype-Phenotype Correlations in Intellectual Disability. Front. Psychiatry 2018, 9, 535. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Levy, D.; Allen, J.; Ye, K.; Ronemus, M.; Lee, Y.H.; Yamrom, B.; Wigler, M. Low load for disruptive mutations in autism genes and their biased transmission. Proc. Natl. Acad. Sci. USA 2015, 112, E5600–E5607. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef]
- Kurki, M.I.; Saarentaus, E.; Pietiläinen, O.; Gormley, P.; Lal, D.; Kerminen, S.; Torniainen-Holm, M.; Hämäläinen, E.; Rahikkala, E.; Keski-Filppula, R.; et al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat. Commun. 2019, 10, 410. [Google Scholar] [CrossRef]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Mitra, I.; Lavillaureix, A.; Yeh, E.; Traglia, M.; Tsang, K.; Bearden, C.E.; Rauen, K.A.; Weiss, L.A. Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. PLoS Genet. 2017, 13, e1006516. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.K.; Bateup, H.S. Modeling Somatic Mutations Associated With Neurodevelopmental Disorders in Human Brain Organoids. Front. Mol. Neurosci. 2022, 14, 341. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef]
- Avni, E.; Ben-Itzchak, E.; Zachor, D.A. The Presence of Comorbid ADHD and Anxiety Symptoms in Autism Spectrum Disorder: Clinical Presentation and Predictors. Front. Psychiatry 2018, 9, 717. [Google Scholar] [CrossRef] [PubMed]
- Tărlungeanu, D.C.; Novarino, G. Genomics in neurodevelopmental disorders: An avenue to personalized medicine. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef]
- Cristino, A.S.; Williams, S.M.; Hawi, Z.; An, J.Y.; Bellgrove, M.A.; Schwartz, C.E.; Costa, L.D.F.; Claudianos, C. Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol. Psychiatry 2013, 19, 294–301. [Google Scholar] [CrossRef]
- Hormozdiari, F.; Penn, O.; Borenstein, E.; Eichler, E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015, 25, 142–154. [Google Scholar] [CrossRef]
- Mullins, C.; Fishell, G.; Tsien, R.W. Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops. Neuron 2016, 89, 1131–1156. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Megagiannis, P.; Suresh, R.; Rouleau, G.A.; Zhou, Y. Reversibility and therapeutic development for neurodevelopmental disorders, insights from genetic animal models. Adv. Drug Deliv. Rev. 2022, 191, 114562. [Google Scholar] [CrossRef]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. DMM Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Mariano, V.; Achsel, T.; Bagni, C.; Kanellopoulos, A.K. Modelling Learning and Memory in Drosophila to Understand Intellectual Disabilities. Neuroscience 2020, 445, 12–30. [Google Scholar] [CrossRef]
- Vaz, R.; Hofmeister, W.; Lindstrand, A. Zebrafish models of neurodevelopmental disorders: Limitations and benefits of current tools and techniques. Int. J. Mol. Sci. 2019, 20, 1296. [Google Scholar] [CrossRef]
- Kim, Y.; Park, Y.; Hwang, J.; Kwack, K. Comparative genomic analysis of the human and nematode caenorhabditis elegans uncovers potential reproductive genes and disease associations in humans. Physiol. Genom. 2018, 50, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Bessa, C.; Maciel, P.; Rodrigues, A.J. Using C. Elegans to decipher the cellular and molecular mechanisms underlying neurodevelopmental disorders. Mol. Neurobiol. 2013, 48, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Tavares, B.; Santos Lopes, S. The Importance of Zebrafish in Biomedical Research. Acta Med. Port. 2013, 26, 583–592. [Google Scholar] [CrossRef]
- de Abreu, M.S.; Genario, R.; Giacomini, A.C.V.V.; Demin, K.A.; Lakstygal, A.M.; Amstislavskaya, T.G.; Fontana, B.D.; Parker, M.O.; Kalueff, A.V. Zebrafish as a Model of Neurodevelopmental Disorders. Neuroscience 2020, 445, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef]
- Homberg, J.R.; Kyzar, E.J.; Nguyen, M.; Norton, W.H.; Pittman, J.; Poudel, M.K.; Gaikwad, S.; Nakamura, S.; Koshiba, M.; Yamanouchi, H.; et al. Understanding autism and other neurodevelopmental disorders through experimental translational neurobehavioral models. Neurosci. Biobehav. Rev. 2016, 65, 292–312. [Google Scholar] [CrossRef]
- Silverman, J.L.; Ellegood, J. Behavioral and neuroanatomical approaches in models of neurodevelopmental disorders: Opportunities for translation. Curr. Opin. Neurol. 2018, 31, 126–133. [Google Scholar] [CrossRef]
- Aida, T.; Feng, G. The dawn of non-human primate models for neurodevelopmental disorders. Curr. Opin. Genet. Dev. 2020, 65, 160–168. [Google Scholar] [CrossRef]
- Jennings, C.G.; Landman, R.; Zhou, Y.; Sharma, J.; Hyman, J.; Movshon, J.A.; Qiu, Z.; Roberts, A.C.; Roe, A.W.; Wang, X.; et al. Opportunities and challenges in modeling human brain disorders in transgenic primates. Nat. Neurosci. 2016, 19, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 2015, 28, 91–102. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Bear, M.F. The Autistic Neuron: Troubled Translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Sur, M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 2015, 350, aab3897. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. mTOR signaling in epilepsy: Insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 2015, 5, 1–18. [Google Scholar] [CrossRef]
- Borrie, S.C.; Brems, H.; Legius, E.; Bagni, C. Cognitive Dysfunctions in Intellectual Disabilities: The Contributions of the Ras-MAPK and PI3K-AKT-mTOR Pathways. Annu. Rev. Genom. Hum. Genet. 2017, 18, 115–142. [Google Scholar] [CrossRef]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R.; de Vries, P.J. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- Iffland, P.H.; Carson, V.; Bordey, A.; Crino, P.B. GATORopathies: The role of amino acid regulatory gene mutations in epilepsy and cortical malformations. Epilepsia 2019, 60, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Otero, M.G.; Grand, K.; Choi, A.; Graham, J.M.; Young, J.I.; Mackay, J.P. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-Van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef]
- Sim, J.C.H.; White, S.M.; Lockhart, P.J. ARID1B-mediated disorders: Mutations and possible mechanisms. Intractable Rare Dis. Res. 2015, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.R.; Cruz, A.C.P.; Ferrasa, A.; Phan, D.; Herai, R.H.; Muotri, A.R. Genetic variations on SETD5 underlying autistic conditions. Dev. Neurobiol. 2018, 78, 500–518. [Google Scholar] [CrossRef]
- Hevner, R.F.; Shi, L.; Justice, N.; Hsueh, Y.P.; Sheng, M.; Smiga, S.; Bulfone, A.; Goffinet, A.M.; Campagnoni, A.T.; Rubenstein, J.L.R. Tbr1 regulates differentiation of the preplate and layer 6. Neuron 2001, 29, 353–366. [Google Scholar] [CrossRef]
- Notwell, J.H.; Heavner, W.E.; Darbandi, S.F.; Katzman, S.; McKenna, W.L.; Ortiz-Londono, C.F.; Tastad, D.; Eckler, M.J.; Rubenstei, J.L.R.; McConnell, S.K.; et al. TBR1 regulates autism risk genes in the developing neocortex. Genome Res. 2016, 26, 1013–1022. [Google Scholar] [CrossRef]
- Traylor, R.N.; Dobyns, W.B.; Rosenfeld, J.A.; Wheeler, P.; Spence, J.E.; Bandholz, A.M.; Bawle, E.V.; Carmany, E.P.; Powell, C.M.; Hudson, B.; et al. Investigation of TBR1 hemizygosity: Four individuals with 2q24 microdeletions. Mol. Syndromol. 2012, 3, 102–112. [Google Scholar] [CrossRef]
- Huang, T.N.; Chuang, H.C.; Chou, W.H.; Chen, C.Y.; Wang, H.F.; Chou, S.J.; Hsueh, Y.P. Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci. 2014, 17, 240–247. [Google Scholar] [CrossRef]
- Huang, T.N.; Hsueh, Y.P. Brain-specific transcriptional regulator T-brain-1 controls brain wiring and neuronal activity in autism spectrum disorders. Front. Neurosci. 2015, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Kentrup, H.; Becker, W.; Heukelbach, J.; Wilmes, A.; Schürmann, A.; Huppertz, C.; Kainulainen, H.; Joost, H.G. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J. Biol. Chem. 1996, 271, 3488–3495. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Evers, J.M.G.; Laskowski, R.A.; Bertolli, M.; Clayton-Smith, J.; Deshpande, C.; Eason, J.; Elmslie, F.; Flinter, F.; Gardiner, C.; Hurst, J.A.; et al. Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders. Hum. Mol. Genet. 2017, 26, 519–526. [Google Scholar] [CrossRef]
- Guimera, J.; Casas, C.; Estivill, X.; Pritchard, M. Human minibrain homologue (MNBH/DYRK1): Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics 1999, 57, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef]
- Altafaj, X.; Dierssen, M.; Baamonde, C.; Martí, E.; Visa, J.; Guimerà, J.; Oset, M.; González, J.R.; Flórez, J.; Fillat, C.; et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 2001, 10, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50. [Google Scholar] [CrossRef]
- Raveau, M.; Shimohata, A.; Amano, K.; Miyamoto, H.; Yamakawa, K. DYRK1A-haploinsufficiency in mice causes autistic-like features and febrile seizures. Neurobiol. Dis. 2018, 110, 180–191. [Google Scholar] [CrossRef]
- Arranz, J.; Balducci, E.; Arató, K.; Sánchez-Elexpuru, G.; Najas, S.; Parras, A.; Rebollo, E.; Pijuan, I.; Erb, I.; Verde, G.; et al. Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 2019, 127, 210–222. [Google Scholar] [CrossRef]
- Leslie, N.R.; Longy, M. Inherited PTEN mutations and the prediction of phenotype. Semin. Cell Dev. Biol. 2016, 52, 30–38. [Google Scholar] [CrossRef]
- Mingo, J.; Rodríguez-Escudero, I.; Luna, S.; Fernández-Acero, T.; Amo, L.; Jonasson, A.R.; Zori, R.T.; López, J.I.; Molina, M.; Cid, V.J.; et al. A pathogenic role for germline PTEN variants which accumulate into the nucleus. Eur. J. Hum. Genet. 2018, 26, 1180–1187. [Google Scholar] [CrossRef]
- Rademacher, S.; Eickholt, B.J. PTEN in autism and neurodevelopmental disorders. Cold Spring Harb. Perspect. Med. 2019, 9, a036780. [Google Scholar] [CrossRef]
- Shin, S.; Santi, A.; Huang, S. Conditional Pten knockout in parvalbumin- or somatostatin-positive neurons sufficiently leads to autism-related behavioral phenotypes. Mol. Brain 2021, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Clipperton-Allen, A.E.; Page, D.T. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum. Mol. Genet. 2014, 23, 3490–3505. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-Linked Mental Retardation and Autism Are Associated with a Mutation in the NLGN4 Gene, a Member of the Neuroligin Family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Burnstein, R.M.; He, X.; Luce, R.; Furlong, R.; Foltynie, T.; Sykacek, P.; Menon, D.K.; Caldwell, M.A. Gene expression changes in long term expanded human neural progenitor cells passaged by chopping lead to loss of neurogenic potential in vivo. Exp. Neurol. 2007, 204, 512–524. [Google Scholar] [CrossRef]
- Trzaska, K.A.; Kuzhikandathil, E.V.; Rameshwar, P. Specification of a Dopaminergic Phenotype from Adult Human Mesenchymal Stem Cells. Stem Cells 2007, 25, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Wakao, S.; Kitada, M.; Ose, T.; Watabe, H.; Kuroda, Y.; Mitsunaga, K.; Matsuse, D.; Shigemoto, T.; Ito, A.; et al. Autologous mesenchymal stem cell-derived dopaminergic neurons function in parkinsonian macaques. J. Clin. Investig. 2013, 123, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Urrutia, D.N.; Caviedes, P.; Mardones, R.; Minguell, J.J.; Vega-Letter, A.M.; Jofre, C.M. Comparative study of the neural differentiation capacity of mesenchymal stromal cells from different tissue sources: An approach for their use in neural regeneration therapies. PLoS ONE 2019, 14, e0213032. [Google Scholar] [CrossRef] [PubMed]
- Wegmeyer, H.; Bröske, A.M.; Leddin, M.; Kuentzer, K.; Nisslbeck, A.K.; Hupfeld, J.; Wiechmann, K.; Kuhlen, J.; Von Schwerin, C.; Stein, C.; et al. Mesenchymal stromal cell characteristics vary depending on their origin. Stem Cells Dev. 2013, 22, 2606–2618. [Google Scholar] [CrossRef]
- Hermann, A.; List, C.; Habisch, H.J.; Vukicevic, V.; Ehrhart-Bornstein, M.; Brenner, R.; Bernstein, P.; Fickert, S.; Storch, A. Age-dependent neuroectodermal differentiation capacity of human mesenchymal stromal cells: Limitations for autologous cell replacement strategies. Cytotherapy 2010, 12, 17–30. [Google Scholar] [CrossRef]
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Hasegawa, K.; Pomeroy, J.E.; Pera, M.F. Current technology for the derivation of pluripotent stem cell lines from human embryos. Cell Stem Cell 2010, 6, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Faravelli, I.; Cardano, M.; Conti, L. Human pluripotent stem cells as tools for neurodegenerative and neurodevelopmental disease modeling and drug discovery. Expert Opin. Drug Discov. 2015, 10, 615–629. [Google Scholar] [CrossRef]
- Quartier, A.; Courraud, J.; Thi Ha, T.; McGillivray, G.; Isidor, B.; Rose, K.; Drouot, N.; Savidan, M.A.; Feger, C.; Jagline, H.; et al. Novel mutations in NLGN3 causing autism spectrum disorder and cognitive impairment. Hum. Mutat. 2019, 40, 2021–2032. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Lehr, A.W.; Roche, K.W. Neuroligins and Neurodevelopmental Disorders: X-Linked Genetics. Front. Synaptic Neurosci. 2020, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients [1]. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Speed, H.E.; Masiulis, I.; Gibson, J.R.; Powell, C.M. Increased cortical inhibition in Autism-Linked neuroligin-3R451C mice is due in part to loss of endocannabinoid signaling. PLoS ONE 2015, 10, e0140638. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.; Földy, C.; Sharma, M.; Tabuchi, K.; Liu, X.; Shamloo, M.; Malenka, R.C.; Südhof, T.C. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. USA 2011, 108, 13764–13769. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef]
- Garrido, D.; Beretta, S.; Grabrucker, S.; Bauer, H.F.; Bayer, D.; Sala, C.; Verpelli, C.; Roselli, F.; Bockmann, J.; Proepper, C.; et al. Shank2/3 double knockout-based screening of cortical subregions links the retrosplenial area to the loss of social memory in autism spectrum disorders. Mol. Psychiatry 2022, 27, 4994–5006. [Google Scholar] [CrossRef]
- Bystron, I.; Blakemore, C.; Rakic, P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008, 9, 110–122. [Google Scholar] [CrossRef]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, G.; Friedrich, T.; Davis-Turak, J.; Winden, K.; Oldham, M.C.; Gao, F.; Chen, L.; Wang, G.Z.; Luo, R.; Preuss, T.M.; et al. Human-Specific Transcriptional Networks in the Brain. Neuron 2012, 75, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Yuen, R.K.C.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, K.; Okano, H. Modeling neurodevelopment in a dish with pluripotent stem cells. Dev. Growth Differ. 2021, 63, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.C. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006, 16, 132–142. [Google Scholar] [CrossRef]
- Kumar, A.; Pareek, V.; Faiq, M.A.; Ghosh, S.K.; Kumari, C. Adult neurogenesis in humans: A review of basic concepts, history, current research, and clinical implications. Innov. Clin. Neurosci. 2019, 16, 30–37. [Google Scholar]
- Yu, Y.; Gu, S.; Huang, H.; Wen, T. Combination of bFGF, heparin and laminin induce the generation of dopaminergic neurons from rat neural stem cells both in vitro and in vivo. J. Neurol. Sci. 2007, 255, 81–86. [Google Scholar] [CrossRef]
- Taylor, C.J.; Bolton, E.M.; Bradley, J.A. Immunological considerations for embryonic and induced pluripotent stem cell banking. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2312–2322. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Jefford, M.; Moore, R. Improvement of informed consent and the quality of consent documents. Lancet Oncol. 2008, 9, 485–493. [Google Scholar] [CrossRef]
- Huang, C.Y.; Liu, C.L.; Ting, C.Y.; Chiu, Y.T.; Cheng, Y.C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best Practices for Translational Disease Modeling Using Human iPSC-Derived Neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014I, 768391. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ät-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Rohani, L.; Johnson, A.A.; Arnold, A.; Stolzing, A. The aging signature: A hallmark of induced pluripotent stem cells? Aging Cell 2014, 13, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.M.; Hennick, K.M.; Regalado, S.G.; Vogan, J.M.; Zhang, X.; Collins, K.; Hockemeyer, D. Telomere length set point regulation in human pluripotent stem cells critically depends on the shelterin protein TPP1. Mol. Biol. Cell 2020, 31, 2583–2596. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Besusso, D.; Conforti, P.; Cattaneo, E. hiPSCs for predictive modelling of neurodegenerative diseases: Dreaming the possible. Nat. Rev. Neurol. 2021, 17, 381–392. [Google Scholar] [CrossRef]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 2008, 454, 646–650. [Google Scholar] [CrossRef]
- Kim, J.B.; Greber, B.; Arazo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-Induced Pluripotency in Adult Neural Stem Cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.; Zhou, H.; Nadif Kasri, N. Choices for Induction of Pluripotency: Recent Developments in Human Induced Pluripotent Stem Cell Reprogramming Strategies. Stem Cell Rev. Rep. 2016, 12, 54–72. [Google Scholar] [CrossRef]
- BORDET, P. The adenoviruses. Brux. Med. 1960, 40, 701–708. [Google Scholar] [CrossRef]
- Siegl, G.; Bates, R.C.; Berns, K.I.; Carter, B.J.; Kelly, D.C.; Kurstak, E.; Tattersall, P. Characteristics and taxonomy of Parvoviridae. Intervirology 1985, 23, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.H.; Moon, J.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Plank, C.; Scherer, F.; Schillinger, U.; Bergemann, C.; Anton, M. Magnetofection: Enhancing and targeting gene delivery with superparamagnetic nanoparticles and magnetic fields. J. Liposome Res. 2003, 13, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Christodoulou, C.; Gianotti-Sommer, A.; Shen, S.S.; Sailaja, B.S.; Hezroni, H.; Spira, A.; Meshorer, E.; Kotton, D.N.; Mostoslavsky, G. Residual Expression of Reprogramming Factors Affects the Transcriptional Program and Epigenetic Signatures of Induced Pluripotent Stem Cells. PLoS ONE 2012, 7, e51711. [Google Scholar] [CrossRef] [PubMed]
- Kadari, A.; Lu, M.; Li, M.; Sekaran, T.; Thummer, R.P.; Guyette, N.; Chu, V.; Edenhofer, F. Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Res. Ther. 2014, 5, 47. [Google Scholar] [CrossRef]
- Hu, K. All roads lead to induced pluripotent stem cells: The technologies of iPSC generation. Stem Cells Dev. 2014, 23, 1285–1300. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Shao, L.; Wu, W.S. Gene-delivery systems for iPS cell generation. Expert Opin. Biol. Ther. 2010, 10, 231–242. [Google Scholar] [CrossRef]
- Wang, H.; Shayakhmetov, D.M.; Leege, T.; Harkey, M.; Li, Q.; Papayannopoulou, T.; Stamatoyannopolous, G.; Lieber, A. A Capsid-Modified Helper-Dependent Adenovirus Vector Containing the β-Globin Locus Control Region Displays a Nonrandom Integration Pattern and Allows Stable, Erythroid-Specific Gene Expression. J. Virol. 2005, 79, 10999–11013. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Baum, B.J.; Iadarola, M.J.; O’Connell, B.C. Genomic integration and gene expression by a modified adenoviral vector. Nat. Biotechnol. 2000, 18, 176–180. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.; Muruve, D. Immune Responses to Adeno-Associated Virus Vectors. Curr. Gene Ther. 2005, 5, 323–331. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Wolf, S.J.; Samulski, R.J. Delivering transgenic DNA exceeding the carrying capacity of AAV vectors. Methods Mol. Biol. 2016, 1382, 21–39. [Google Scholar] [PubMed]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Bayart, E.; Cohen-Haguenauer, O. Technological Overview of iPS Induction from Human Adult Somatic Cells. Curr. Gene Ther. 2013, 13, 73–92. [Google Scholar] [CrossRef]
- Beers, J.; Linask, K.L.; Chen, J.A.; Siniscalchi, L.I.; Lin, Y.; Zheng, W.; Rao, M.; Chen, G. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep. 2015, 5, 11319. [Google Scholar] [CrossRef]
- Dey, C.; Narayan, G.; Krishna Kumar, H.; Borgohain, M.; Lenka, N. Cell-Penetrating Peptides as a Tool to Deliver Biologically Active Recombinant Proteins to Generate Transgene-Free Induced Pluripotent Stem Cells. Stud. Stem Cells Res. Ther. 2017, 3, 006–015. [Google Scholar] [CrossRef]
- Thier, M.; Münst, B.; Mielke, S.; Edenhofer, F. Cellular reprogramming employing recombinant Sox2 protein. Stem Cells Int. 2012, 2012, 549846. [Google Scholar] [CrossRef]
- Thier, M.; Münst, B.; Edenhofer, F. Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. Int. J. Dev. Biol. 2011, 54, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Armeanu, S.; Lauer, U.M.; Neubert, W.J. Sendai virus vectors as an emerging negative-strand RNA viral vector system. J. Gene Med. 2003, 5, 543–553. [Google Scholar] [CrossRef]
- Hosoya, N.; Miura, T.; Kawana-Tachikawa, A.; Koibuchi, T.; Shioda, T.; Odawara, T.; Nakamura, T.; Kitamura, Y.; Kano, M.; Kato, A.; et al. Comparison between Sendai virus and adenovirus vectors to transduce HIV-1 genes into human dendritic cells. J. Med. Virol. 2008, 80, 373–382. [Google Scholar] [CrossRef]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef]
- Nishimura, K.; Ohtaka, M.; Takada, H.; Kurisaki, A.; Tran, N.V.K.; Tran, Y.T.H.; Hisatake, K.; Sano, M.; Nakanishi, M. Simple and effective generation of transgene-free induced pluripotent stem cells using an auto-erasable Sendai virus vector responding to microRNA-302. Stem Cell Res. 2017, 23, 13–19. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010, 394, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Plews, J.R.; Li, J.L.; Jones, M.; Moore, H.D.; Mason, C.; Andrews, P.W.; Na, J. Activation of pluripotency genes in human fibroblast cells by a novel mRNA based approach. PLoS ONE 2010, 5, e14397. [Google Scholar] [CrossRef]
- Steichen, C.; Luce, E.; Maluenda, J.; Tosca, L.; Moreno-Gimeno, I.; Desterke, C.; Dianat, N.; Goulinet-Mainot, S.; Awan-Toor, S.; Burks, D.; et al. Messenger RNA- Versus Retrovirus-Based Induced Pluripotent Stem Cell Reprogramming Strategies: Analysis of Genomic Integrity. Stem Cells Transl. Med. 2014, 3, 686–691. [Google Scholar] [CrossRef]
- Drews, K.; Tavernier, G.; Demeester, J.; Lehrach, H.; De Smedt, S.C.; Rejman, J.; Adjaye, J. The cytotoxic and immunogenic hurdles associated with non-viral mRNA-mediated reprogramming of human fibroblasts. Biomaterials 2012, 33, 4059–4068. [Google Scholar] [CrossRef]
- Angel, M.; Yanik, M.F. Innate immune suppression enables frequent transfection with RNA encoding reprogramming proteins. PLoS ONE 2010, 5, e11756. [Google Scholar] [CrossRef] [Green Version]
- Preskey, D.; Allison, T.F.; Jones, M.; Mamchaoui, K.; Unger, C. Synthetically modified mRNA for efficient and fast human iPS cell generation and direct transdifferentiation to myoblasts. Biochem. Biophys. Res. Commun. 2016, 473, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Rohani, L.; Fabian, C.; Holland, H.; Naaldijk, Y.; Dressel, R.; Löffler-Wirth, H.; Binder, H.; Arnold, A.; Stolzing, A. Generation of human induced pluripotent stem cells using non-synthetic mRNA. Stem Cell Res. 2016, 16, 662–672. [Google Scholar] [CrossRef]
- Choi, H.Y.; Lee, T.J.; Yang, G.M.; Oh, J.; Won, J.; Han, J.; Jeong, G.J.; Kim, J.; Kim, J.H.; Kim, B.S.; et al. Efficient mRNA delivery with graphene oxide-polyethylenimine for generation of footprint-free human induced pluripotent stem cells. J. Control. Release 2016, 235, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Poleganov, M.A.; Eminli, S.; Beissert, T.; Herz, S.; Moon, J.; Goldmann, J.; Beyer, A.; Heck, R.; Burkhart, I.; Barea Roldan, D.; et al. Efficient Reprogramming of Human Fibroblasts and Blood-Derived Endothelial Progenitor Cells Using Nonmodified RNA for Reprogramming and Immune Evasion. Hum. Gene Ther. 2015, 26, 751–766. [Google Scholar] [CrossRef]
- Kogut, I.; McCarthy, S.M.; Pavlova, M.; Astling, D.P.; Chen, X.; Jakimenko, A.; Jones, K.L.; Getahun, A.; Cambier, J.C.; Pasmooij, A.M.G.; et al. High-efficiency RNA-based reprogramming of human primary fibroblasts. Nat. Commun. 2018, 9, 745. [Google Scholar] [CrossRef]
- Heng, B.C.; Heinimann, K.; Miny, P.; Iezzi, G.; Glatz, K.; Scherberich, A.; Zulewski, H.; Fussenegger, M. MRNA transfection-based, feeder-free, induced pluripotent stem cells derived from adipose tissue of a 50-year-old patient. Metab. Eng. 2013, 18, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Ni, Y.; Wang, J.; Guo, X. Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci. Rep. 2012, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Durruthy, J.D.; Sebastiano, V. Derivation of GMP-compliant integration-free hiPSCs using modified mRNAs. Methods Mol. Biol. 2015, 1283, 31–42. [Google Scholar] [CrossRef]
- Maurisse, R.; De Semir, D.; Emamekhoo, H.; Bedayat, B.; Abdolmohammadi, A.; Parsi, H.; Gruenert, D.C. Comparative transfection of DNA into primary and transformed mammalian cells from different lineages. BMC Biotechnol. 2010, 10, 1–9. [Google Scholar] [CrossRef]
- Montserrat, N.; Garreta, E.; González, F.; Gutiérrez, J.; Eguizábal, C.; Ramos, V.; Borrós, S.; Belmonte, J.C.I. Simple generation of human induced pluripotent stem cells using poly-β-amino esters as the non-viral gene delivery system. J. Biol. Chem. 2011, 286, 12417–12428. [Google Scholar] [CrossRef] [Green Version]
- Chabot, S.; Orio, J.; Schmeer, M.; Schleef, M.; Golzio, M.; Teissié, J. Minicircle DNA electrotransfer for efficient tissue-targeted gene delivery. Gene Ther. 2013, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Kong, L.; Zhu, S. Reprogramming cell fates by small molecules. Protein Cell 2017, 8, 328–348. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia Enhances the Generation of Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef]
- Maucksch, C.; Bohla, A.; Hoffmann, F.; Schleef, M.; Aneja, M.K.; Elfinger, M.; Hartl, D.; Rudolph, C. Transgene expression of transfected supercoiled plasmid DNA concatemers in mammalian cells. J. Gene Med. 2009, 11, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; He, C.Y.; Ehrhardt, A.; Kay, M.A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 2003, 8, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Galla, M.; Schambach, A.; Falk, C.S.; Maetzig, T.; Kuehle, J.; Lange, K.; Zychlinski, D.; Heinz, N.; Brugman, M.H.; Göhring, G.; et al. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011, 39, 7147–7160. [Google Scholar] [CrossRef]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef]
- Kumar, D.; Talluri, T.R.; Anand, T.; Kues, W.A. Transposon-based reprogramming to induced pluripotency. Histol. Histopathol. 2015, 30, 1397–1409. [Google Scholar] [CrossRef]
- Scherer, F.; Anton, M.; Schillinger, U.; Henke, J.; Bergemann, C.; Krüger, A.; Gänsbacher, B.; Plank, C. Magnetofection: Enhancing and targeting gene delivery by magnetic force in vitro and in vivo. Gene Ther. 2002, 9, 102–109. [Google Scholar] [CrossRef]
- Park, H.Y.; Noh, E.H.; Chung, H.M.; Kang, M.J.; Kim, E.Y.; Park, S.P. Efficient Generation of Virus-Free iPS Cells Using Liposomal Magnetofection. PLoS ONE 2012, 7, e45812. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Taniguchi, Y.; Senda, S.; Takizawa, N.; Ichisaka, T.; Asano, K.; Morizane, A.; Doi, D.; Takahashi, J.; Nishizawa, M.; et al. A novel efficient feeder-Free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 2014, 4, 3594. [Google Scholar] [CrossRef]
- Ji, J.; Sharma, V.; Qi, S.; Guarch, M.E.; Zhao, P.; Luo, Z.; Fan, W.; Wang, Y.; Mbabaali, F.; Neculai, D.; et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 44–51. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.N.; Guoynes, C.; Cho, J.; Hao, J.; Lutfy, K.; Hong, Y. Rapid generation of sub-type, region-specific neurons and neural networks from human pluripotent stem cell-derived neurospheres. Stem Cell Res. 2015, 15, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Son, E.Y.; Ichida, J.K.; Wainger, B.J.; Toma, J.S.; Rafuse, V.F.; Woolf, C.J.; Eggan, K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 2011, 9, 205–218. [Google Scholar] [CrossRef]
- Hester, M.E.; Murtha, M.J.; Song, S.; Rao, M.; Miranda, C.J.; Meyer, K.; Tian, J.; Boulting, G.; Schaffer, D.V.; Zhu, M.X.; et al. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 2011, 19, 1905–1912. [Google Scholar] [CrossRef]
- Santos, D.P.; Kiskinis, E. Generation of spinal motor neurons from human pluripotent stem cells. Methods Mol. Biol. 2017, 1538, 53–66. [Google Scholar] [PubMed]
- Solomon, E.; Davis-Anderson, K.; Hovde, B.; Micheva-Viteva, S.; Harris, J.F.; Twary, S.; Iyer, R. Global transcriptome profile of the developmental principles of in vitro iPSC-to-motor neuron differentiation. BMC Mol. Cell Biol. 2021, 22, 13. [Google Scholar] [CrossRef]
- Imamura, K.; Kawaguchi, J.; Shu, T.; Inoue, H. Generation of Motor Neurons from Human ESCs/iPSCs Using Sendai Virus Vectors. Methods Mol. Biol. 2021, 2352, 127–132. [Google Scholar]
- Amimoto, N.; Nishimura, K.; Shimohama, S.; Takata, K. Generation of striatal neurons from human induced pluripotent stem cells by controlling extrinsic signals with small molecules. Stem Cell Res. 2021, 55, 102486. [Google Scholar] [CrossRef]
- Dhingra, A.; Täger, J.; Bressan, E.; Rodriguez-Nieto, S.; Bedi, M.S.; Bröer, S.; Sadikoglou, E.; Fernandes, N.; Castillo-Lizardo, M.; Rizzu, P.; et al. Automated production of human induced pluripotent stem cell-derived cortical and dopaminergic neurons with integrated live-cell monitoring. J. Vis. Exp. 2020, 2020, 1–29. [Google Scholar] [CrossRef]
- Silva, M.C.; Nandi, G.; Haggarty, S.J. Differentiation of Human Induced Pluripotent Stem Cells into Cortical Neurons to Advance Precision Medicine. Methods Mol. Biol. 2022, 2429, 143–174. [Google Scholar]
- Lopez-Lengowski, K.; Kathuria, A.; Gerlovin, K.; Karmacharya, R. Co-culturing microglia and cortical neurons differentiated from human induced pluripotent stem cells. J. Vis. Exp. 2021, 2021, e62480. [Google Scholar] [CrossRef]
- Ho, S.M.; Hartley, B.J.; TCW, J.; Beaumont, M.; Stafford, K.; Slesinger, P.A.; Brennand, K.J. Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 2016, 101, 113–124. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Sauvey, C.; Yao, L.; Zarnowska, E.D.; Zhang, S.C. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat. Protoc. 2013, 8, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Tofoli, F.A.; Semeano, A.T.S.; Oliveira-Giacomelli, Á.; Gonçalves, M.C.B.; Ferrari, M.F.R.; Veiga Pereira, L.; Ulrich, H. Midbrain dopaminergic neurons differentiated from human-induced pluripotent stem cells. Methods Mol. Biol. 2019, 1919, 97–118. [Google Scholar]
- Corti, S.; Bonjean, R.; Legier, T.; Rattier, D.; Melon, C.; Salin, P.; Toso, E.A.; Kyba, M.; Kerkerian-Le Goff, L.; Maina, F.; et al. Enhanced differentiation of human induced pluripotent stem cells toward the midbrain dopaminergic neuron lineage through GLYPICAN-4 downregulation. Stem Cells Transl. Med. 2021, 10, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Mahajani, S.; Raina, A.; Fokken, C.; Kügler, S.; Bähr, M. Homogenous generation of dopaminergic neurons from multiple hiPSC lines by transient expression of transcription factors. Cell Death Dis. 2019, 10, 898. [Google Scholar] [CrossRef]
- Xue, Y.; Zhan, X.; Sun, S.; Karuppagounder, S.S.; Xia, S.; Dawson, V.L.; Dawson, T.M.; Laterra, J.; Zhang, J.; Ying, M. Synthetic mRNAs Drive Highly Efficient iPS Cell Differentiation to Dopaminergic Neurons. Stem Cells Transl. Med. 2019, 8, 112–123. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, H.; Zhong, P.; Yan, Z.; Chen, S.; Feng, J. Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 2016, 21, 62–70. [Google Scholar] [CrossRef]
- Jansch, C.; Ziegler, G.C.; Forero, A.; Gredy, S.; Wäldchen, S.; Vitale, M.R.; Svirin, E.; Zöller, J.E.M.; Waider, J.; Günther, K.; et al. Serotonin-specific neurons differentiated from human iPSCs form distinct subtypes with synaptic protein assembly. J. Neural Transm. 2021, 128, 225–241. [Google Scholar] [CrossRef]
- Neureiter, A.; Eberhardt, E.; Lampert, A. Differentiation of iPS-Cells into Peripheral Sensory Neurons. Methods Mol. Biol. 2022, 2429, 175–188. [Google Scholar] [PubMed]
- Gupta, S.; Sivalingam, D.; Hain, S.; Makkar, C.; Sosa, E.; Clark, A.; Butler, S.J. Deriving Dorsal Spinal Sensory Interneurons from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 390–405. [Google Scholar] [CrossRef]
- Cantor, E.L.; Shen, F.; Jiang, G.; Tan, Z.; Cunningham, G.M.; Wu, X.; Philips, S.; Schneider, B.P. Passage number affects differentiation of sensory neurons from human induced pluripotent stem cells. Sci. Rep. 2022, 12, 15869. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Topol, A.; English, J.A.; Flaherty, E.; Rajarajan, P.; Hartley, B.J.; Gupta, S.; Desland, F.; Zhu, S.; Goff, T.; Friedman, L.; et al. Increased abundance of translation machinery in stem cell–derived neural progenitor cells from four schizophrenia patients. Transl. Psychiatry 2015, 5, e662. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of Gene Knockout Mice. Curr. Protoc. Cell Biol. 2009, 44, 19.12.1–19.12.17. [Google Scholar] [CrossRef]
- Tong, C.; Huang, G.; Ashton, C.; Li, P.; Ying, Q.L. Generating gene knockout rats by homologous recombination in embryonic stem cells. Nat. Protoc. 2011, 6, 827–844. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.I.; Muguruma, K.; et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Bañuelos, C.A.; Banáth, J.P.; MacPhail, S.H.; Zhao, J.; Eaves, C.A.; O’Connor, M.D.; Lansdorp, P.M.; Olive, P.L. Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair 2008, 7, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Tesson, L.; Usal, C.; Ménoret, S.; Leung, E.; Niles, B.J.; Remy, S.; Santiago, Y.; Vincent, A.I.; Meng, X.; Zhang, L.; et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 2011, 29, 695–696. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Soldner, F.; Laganière, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.K.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, P.; Mazurek, S.; Kołodziejczak, I.; Wiznerowicz, M. Gene delivery methods and genome editing of human pluripotent stem cells. Rep. Pract. Oncol. Radiother. 2019, 24, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Korecka, J.A.; Talbot, S.; Osborn, T.M.; de Leeuw, S.M.; Levy, S.A.; Ferrari, E.J.; Moskites, A.; Atkinson, E.; Jodelka, F.M.; Hinrich, A.J.; et al. Neurite Collapse and Altered ER Ca 2+ Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem Cell Rep. 2019, 12, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector article. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, A.; Diels, A.; Reumers, J.; Van Hoorde, K.; Van den Wyngaert, I.; van Outryve d’Ydewalle, C.; De Bondt, A.; Kuijlaars, J.; De Muynck, L.; De Hoogt, R.; et al. Genetically Engineered iPSC-Derived FTDP-17 MAPT Neurons Display Mutation-Specific Neurodegenerative and Neurodevelopmental Phenotypes. Stem Cell Rep. 2018, 11, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.W.; Haidet-Phillips, A.M.; Pham, J.T.; Lee, Y.; Huo, Y.; Tienari, P.J.; Maragakis, N.J.; Sattler, R.; Rothstein, J.D. Generation of GFAP::GFP astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia 2016, 64, 63–75. [Google Scholar] [CrossRef]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. eBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef]
- Cerbini, T.; Funahashi, R.; Luo, Y.; Liu, C.; Park, K.; Rao, M.; Malik, N.; Zou, J. Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines. PLoS ONE 2015, 10, e0116032. [Google Scholar] [CrossRef]
- Pei, Y.; Sierra, G.; Sivapatham, R.; Swistowski, A.; Rao, M.S.; Zeng, X. A platform for rapid generation of single and multiplexed reporters in human iPSC lines. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Luo, Y.; Rao, M.; Zou, J. Generation of GFP reporter human induced pluripotent stem cells using AAVS1 safe harbor transcription activator-like effector nuclease. Curr. Protoc. Stem Cell Biol. 2014, 2014, 5a–7. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef]
- Fischer, J.; Heide, M.; Huttner, W.B. Genetic Modification of Brain Organoids. Front. Cell. Neurosci. 2019, 13, 558. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mimitou, E.P.; Cheng, A.; Montalbano, A.; Hao, S.; Stoeckius, M.; Legut, M.; Roush, T.; Herrera, A.; Papalexi, E.; Ouyang, Z.; et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 2019, 16, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef]
- Hendriks, W.T.; Warren, C.R.; Cowan, C.A. Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell Stem Cell 2016, 18, 53–65. [Google Scholar] [CrossRef]
- Kim, D.S.; Joel Ross, P.; Zaslavsky, K.; Ellis, J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front. Cell. Neurosci. 2014, 8, 109. [Google Scholar] [CrossRef]
- Plant, A.L.; Locascio, L.E.; May, W.E.; Gallagher, P.D. Improved reproducibility by assuring confidence in measurements in biomedical research. Nat. Methods 2014, 11, 895–898. [Google Scholar] [CrossRef]
- Crook, J.M.; Hei, D.; Stacey, G. The international stem cell banking initiative (ISCBI): Raising standards to bank on. Vitr. Cell. Dev. Biol.-Anim. 2010, 46, 169–172. [Google Scholar] [CrossRef]
- Li, J.; Song, W.; Pan, G.; Zhou, J. Advances in understanding the cell types and approaches used for generating induced pluripotent stem cells. J. Hematol. Oncol. 2014, 7, 1–18. [Google Scholar] [CrossRef]
- Shofuda, T.; Fukusumi, H.; Kanematsu, D.; Yamamoto, A.; Yamasaki, M.; Arita, N.; Kanemura, Y. A method for efficiently generating neurospheres from human-induced pluripotent stem cells using microsphere arrays. Neuroreport 2013, 24, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Stover, A.E.; Brick, D.J.; Nethercott, H.E.; Banuelos, M.G.; Sun, L.; O’Dowd, D.K.; Schwartz, P.H. Process-based expansion and neural differentiation of human pluripotent stem cells for transplantation and disease modeling. J. Neurosci. Res. 2013, 91, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef]
- Streeter, I.; Harrison, P.W.; Faulconbridge, A.; Consortium, T.H.S.; Flicek, P.; Parkinson, H.; Clarke, L. The human-induced pluripotent stem cell initiative-Data resources for cellular genetics. Nucleic Acids Res. 2017, 45, D691–D697. [Google Scholar] [CrossRef] [PubMed]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef]
- D’Antonio, M.; Benaglio, P.; Jakubosky, D.; Greenwald, W.W.; Matsui, H.; Donovan, M.K.R.; Li, H.; Smith, E.N.; D’Antonio-Chronowska, A.; Frazer, K.A. Insights into the Mutational Burden of Human Induced Pluripotent Stem Cells from an Integrative Multi-Omics Approach. Cell Rep. 2018, 24, 883–894. [Google Scholar] [CrossRef]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.D.L.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human ipscs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar] [CrossRef]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Benvenisty, N. Epigenetic aberrations in human pluripotent stem cells. EMBO J. 2019, 38, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Hongguang, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; Ogmalley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef]
- Roost, M.S.; Slieker, R.C.; Bialecka, M.; Van Iperen, L.; Gomes Fernandes, M.M.; He, N.; Suchiman, H.E.D.; Szuhai, K.; Carlotti, F.; De Koning, E.J.P.; et al. DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat. Commun. 2017, 8, 908. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–17. [Google Scholar] [CrossRef]
- Chen, H.; Kaitlyn Victor, A.; Klein, J.; Tacer, K.F.; Tai, D.J.C.; de Esch, C.; Nuttle, A.; Temirov, J.; Burnett, L.C.; Rosenbaum, M.; et al. Loss of MAGEL2 in prader-willi syndrome leads to decreased secretory granule and neuropeptide production. JCI Insight. 2020, 5, e138576. [Google Scholar] [CrossRef]
- Yang, L.; Shu, X.; Mao, S.; Wang, Y.; Du, X.; Zou, C. Genotype–phenotype correlations in angelman syndrome. Genes 2021, 12, 987. [Google Scholar] [CrossRef]
- Pólvora-Brandão, D.; Joaquim, M.; Godinho, I.; Aprile, D.; Álvaro, A.R.; Onofre, I.; Raposo, A.C.; Pereira de Almeida, L.; Duarte, S.T.; da Rocha, S.T. Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet. 2018, 27, 3999–4011. [Google Scholar] [CrossRef]
- Pick, M.; Stelzer, Y.; Bar-Nur, O.; Mayshar, Y.; Eden, A.; Benvenisty, N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells 2009, 27, 2686–2690. [Google Scholar] [CrossRef]
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Müller, F.J.; Wang, Y.C.; Boscolo, F.S.; et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 2012, 10, 620–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannesson, B.; Sagi, I.; Gore, A.; Paull, D.; Yamada, M.; Golan-Lev, T.; Li, Z.; LeDuc, C.; Shen, Y.; Stern, S.; et al. Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell 2014, 15, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Schachter, M.; Eldar-Geva, T.; Benvenisty, N. Large-Scale Analysis of Loss of Imprinting in Human Pluripotent Stem Cells. Cell Rep. 2017, 19, 957–968. [Google Scholar] [CrossRef]
- Kuwajima, T.; Nishimura, I.; Yoshikawa, K. Cellular/Molecular Necdin Promotes GABAergic Neuron Differentiation in Cooperation with Dlx Homeodomain Proteins. J. Neurosci. 2006, 26, 5383–5392. [Google Scholar] [CrossRef]
- Dimitropoulos, A.; Fearer, I.D.; Roof, E.; Stone, W.; Butler, M.G.; Sutcliffe, J.; Thompson, T. Appetitive behavior, compulsivity, and neurochemistry in Prader-Willi syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Aspects Med. 2013, 34, 826–840. [Google Scholar] [CrossRef]
- Lessing, D.; Anguera, M.C.; Lee, J.T. X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu. Rev. Genom. Hum. Genet. 2013, 14, 85–110. [Google Scholar] [CrossRef]
- Bruck, T.; Benvenisty, N. Meta-analysis of the heterogeneity of X chromosome inactivation in human pluripotent stem cells. Stem Cell Res. 2011, 6, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; De la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef]
- Marks, H.; Chow, J.C.; Denissov, S.; Françoijs, K.J.; Brockdorff, N.; Heard, E.; Stunnenberg, H.G. High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 2009, 19, 1361–1373. [Google Scholar] [CrossRef] [Green Version]
- Tchieu, J.; Kuoy, E.; Chin, M.H.; Trinh, H.; Patterson, M.; Sherman, S.P.; Aimiuwu, O.; Lindgren, A.; Hakimian, S.; Zack, J.A.; et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 2010, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Pomp, O.; Dreesen, O.; Leong, D.F.M.; Meller-Pomp, O.; Tan, T.T.; Zhou, F.; Colman, A. Unexpected X chromosome skewing during culture and reprogramming of human somatic cells can be alleviated by exogenous telomerase. Cell Stem Cell 2011, 9, 156–165. [Google Scholar] [CrossRef]
- Mekhoubad, S.; Bock, C.; De Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef]
- Yue, M.; Charles Richard, J.L.; Yamada, N.; Ogawa, A.; Ogawa, Y. Quick fluorescent in situ hybridization protocol for Xist RNA combined with immunofluorescence of histone modification in X-chromosome inactivation. J. Vis. Exp. 2014, 93, e52053. [Google Scholar] [CrossRef]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.H.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Theka, I.; Caiazzo, M.; Dvoretskova, E.; Leo, D.; Ungaro, F.; Curreli, S.; Managò, F.; Dell’Anno, M.T.; Pezzoli, G.; Gainetdinov, R.R.; et al. Rapid Generation of Functional Dopaminergic Neurons From Human Induced Pluripotent Stem Cells Through a Single-Step Procedure Using Cell Lineage Transcription Factors. Stem Cells Transl. Med. 2013, 2, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Vadodaria, K.C.; Mertens, J.; Paquola, A.; Bardy, C.; Li, X.; Jappelli, R.; Fung, L.; Marchetto, M.C.; Hamm, M.; Gorris, M.; et al. Generation of functional human serotonergic neurons from fibroblasts. Mol. Psychiatry 2016, 21, 49–61. [Google Scholar] [CrossRef]
- Goto, K.; Imamura, K.; Komatsu, K.; Mitani, K.; Aiba, K.; Nakatsuji, N.; Inoue, M.; Kawata, A.; Yamashita, H.; Takahashi, R.; et al. Simple Derivation of Spinal Motor Neurons from ESCs/iPSCs Using Sendai Virus Vectors. Mol. Ther. Methods Clin. Dev. 2017, 4, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.L.; Cui, Q.L.; Schambach, A.; Kim, K.P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef]
- Quadrato, G.; Brown, J.; Arlotta, P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med. 2016, 22, 1220–1228. [Google Scholar] [CrossRef]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef]
- McCauley, H.A.; Wells, J.M. Pluripotent stem cell-derived organoids: Using principles of developmental biology to grow human tissues in a dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Xiang, Y.; Cakir, B.; Park, I.H. Deconstructing and reconstructing the human brain with regionally specified brain organoids. Semin. Cell Dev. Biol. 2021, 111, 40–51. [Google Scholar] [CrossRef]
- Tanaka, Y.; Cakir, B.; Xiang, Y.; Sullivan, G.J.; Park, I.H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020, 30, 1682–1689.e3. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef]
- Chen, H.; Jin, X.; Li, T.; Ye, Z. Brain organoids: Establishment and application. Front. Cell Dev. Biol. 2022, 10, 1029873. [Google Scholar] [CrossRef]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.C.; Beltrao-Braga, P.C.; Trujillo, C.A.; Mendes, A.P.D.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef]
- Grunwald, L.M.; Stock, R.; Haag, K.; Buckenmaier, S.; Eberle, M.C.; Wildgruber, D.; Storchak, H.; Kriebel, M.; Weißgraeber, S.; Mathew, L.; et al. Comparative characterization of human induced pluripotent stem cells (hiPSC) derived from patients with schizophrenia and autism. Transl. Psychiatry 2019, 9, 179. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef]
- Russo, F.B.; Freitas, B.C.; Pignatari, G.C.; Fernandes, I.R.; Sebat, J.; Muotri, A.R.; Beltrão-Braga, P.C.B. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol. Psychiatry 2018, 83, 569–578. [Google Scholar] [CrossRef]
- Kathuria, A.; Nowosiad, P.; Jagasia, R.; Aigner, S.; Taylor, R.D.; Andreae, L.C.; Gatford, N.J.F.; Lucchesi, W.; Srivastava, D.P.; Price, J. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 2018, 23, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Chen, S.; Chen, X.; Zheng, J.; Xu, Z.; Doostparast Torshizi, A.; Gong, S.; Chen, Q.; Ma, X.; Yu, J.; et al. Uncovering the functional link between SHANK3 deletions and deficiency in neurodevelopment using iPSC-derived human neurons. Front. Neuroanat. 2019, 13, 23. [Google Scholar] [CrossRef]
- Lutz, A.K.; Pérez Arévalo, A.; Ioannidis, V.; Stirmlinger, N.; Demestre, M.; Delorme, R.; Bourgeron, T.; Boeckers, T.M. SHANK2 Mutations Result in Dysregulation of the ERK1/2 Pathway in Human Induced Pluripotent Stem Cells-Derived Neurons and Shank2(−/−) Mice. Front. Mol. Neurosci. 2021, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, K.; Zhang, W.B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Avazzadeh, S.; McDonagh, K.; Reilly, J.; Wang, Y.; Boomkamp, S.D.; McInerney, V.; Krawczyk, J.; Fitzgerald, J.; Feerick, N.; O’Sullivan, M.; et al. Increased Ca2+ signaling in NRXN1α +/- neurons derived from ASD induced pluripotent stem cells. Mol. Autism 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Avazzadeh, S.; Quinlan, L.R.; Reilly, J.; McDonagh, K.; Jalali, A.; Wang, Y.; McInerney, V.; Krawczyk, J.; Ding, Y.; Fitzgerald, J.; et al. NRXN1α+/- is associated with increased excitability in ASD iPSC-derived neurons. BMC Neurosci. 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; O’Sullivan, M.L.; Sanchez, C.A.; Hwang, M.; Israel, M.A.; Brennand, K.; Deerinck, T.J.; Goldstein, L.S.B.; Gage, F.H.; Ellisman, M.H.; et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 3005–3010. [Google Scholar] [CrossRef]
- Lim, C.S.; Kim, M.J.; Choi, J.E.; Islam, M.A.; Lee, Y.K.; Xiong, Y.; Shim, K.W.; Yang, J.; Lee, R.U.; Lee, J.; et al. Dysfunction of NMDA receptors in neuronal models of an autism spectrum disorder patient with a DSCAM mutation and in Dscam-knockout mice. Mol. Psychiatry 2021, 26, 7538–7549. [Google Scholar] [CrossRef]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5 -/+ or EHMT2 -/+ human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. eLife 2019, 8, e40092. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol. Autism 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Wagh, V.; Reis, S.A.; Erdin, S.; Beauchamp, R.L.; Shaikh, G.; Talkowski, M.; Thiele, E.; Sheridan, S.D.; Haggarty, S.J.; et al. TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol. Autism 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Z.; Tan, L.; Wang, Y.; Lu, C.; Chen, R.; Zhang, S.; Gao, Y.; Liu, Y.; Yin, Y.; et al. Correcting miR92a-vGAT-Mediated GABAergic Dysfunctions Rescues Human Tau-Induced Anxiety in Mice. Mol. Ther. 2017, 25, 140–152. [Google Scholar] [CrossRef]
- Zhuang, W.; Liu, H.; He, Z.; Ju, J.; Gao, Q.; Shan, Z.; Lei, L. miR-92a-2-5p Regulates the Proliferation and Differentiation of ASD-Derived Neural Progenitor Cells. Curr. Issues Mol. Biol. 2022, 44, 2431–2442. [Google Scholar] [CrossRef]
- Jung, E.-M.; Moffat, J.J.; Liu, J.; Dravid, S.M.; Gurumurthy, C.B.; Kim, W.-Y. Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci. 2017, 20, 1694–1707. [Google Scholar] [CrossRef]
- Smith, A.L.; Jung, E.M.; Jeon, B.T.; Kim, W.Y. Arid1b haploinsufficiency in parvalbumin- or somatostatin-expressing interneurons leads to distinct ASD-like and ID-like behavior. Sci. Rep. 2020, 10, 7834. [Google Scholar] [CrossRef]
- Celen, C.; Chuang, J.C.; Luo, X.; Nijem, N.; Walker, A.K.; Chen, F.; Zhang, S.; Chung, A.S.; Nguyen, L.H.; Nassour, I.; et al. Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. eLife 2017, 6, e25730. [Google Scholar] [CrossRef]
- Ellegood, J.; Petkova, S.P.; Kinman, A.; Qiu, L.R.; Adhikari, A.; Wade, A.A.; Fernandes, D.; Lindenmaier, Z.; Creighton, A.; Nutter, L.M.J.; et al. Neuroanatomy and behavior in mice with a haploinsufficiency of AT-rich interactive domain 1B (ARID1B) throughout development. Mol. Autism 2021, 12, 25. [Google Scholar] [CrossRef]
- Shibutani, M.; Horii, T.; Shoji, H.; Morita, S.; Kimura, M.; Terawaki, N.; Miyakawa, T.; Hatada, I. Arid1b haploinsufficiency causes abnormal brain gene expression and autism-related behaviors in mice. Int. J. Mol. Sci. 2017, 18, 1872. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, D.; Cho, Y.; Kim, K.; Roh, J.D.; Kim, Y.; Yang, E.; Kim, S.S.; Ahn, S.; Kim, H.; et al. Early postnatal serotonin modulation prevents adult-stage deficits in Arid1b-deficient mice through synaptic transcriptional reprogramming. Nat. Commun. 2022, 13, 5051. [Google Scholar] [CrossRef] [PubMed]
- Ka, M.; Chopra, D.A.; Dravid, S.M.; Kim, W.Y. Essential Roles for ARID1B in Dendritic Arborization and Spine Morphology of Developing Pyramidal Neurons. J. Neurosci. 2016, 36, 2723–2742. [Google Scholar] [CrossRef]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-related syndrome: A comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Mok, R.S.F.; Zhang, W.; Sheikh, T.I.; Pradeepan, K.; Fernandes, I.R.; DeJong, L.C.; Benigno, G.; Hildebrandt, M.R.; Mufteev, M.; Rodrigues, D.C.; et al. Wide spectrum of neuronal and network phenotypes in human stem cell-derived excitatory neurons with Rett syndrome-associated MECP2 mutations. Transl. Psychiatry 2022, 12, 450. [Google Scholar] [CrossRef]
- Perego, S.; Alari, V.; Pietra, G.; Lamperti, A.; Vimercati, A.; Camporeale, N.; Garzo, M.; Cogliati, F.; Milani, D.; Vignoli, A.; et al. Modeling RTT Syndrome by iPSC-Derived Neurons from Male and Female Patients with Heterogeneously Severe Hot-Spot MECP2 Variants. Int. J. Mol. Sci. 2022, 23, 4491. [Google Scholar] [CrossRef]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J.; et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef] [PubMed]
- Patriarchi, T.; Amabile, S.; Frullanti, E.; Landucci, E.; Lo Rizzo, C.; Ariani, F.; Costa, M.; Olimpico, F.; Hell, J.W.; Vaccarino, F.M.; et al. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1 patients and in foxg1 mice. Eur. J. Hum. Genet. 2016, 24, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Schaevitz, L.R.; Gómez, N.B.; Zhen, D.P.; Berger-Sweeney, J.E. MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Genes Brain Behav. 2013, 12, 732–740. [Google Scholar] [CrossRef]
- Tai, D.J.C.; Liu, Y.C.; Hsu, W.L.; Ma, Y.L.; Cheng, S.J.; Liu, S.Y.; Lee, E.H.Y. MeCP2 SUMOylation rescues Mecp2-mutant-induced behavioural deficits in a mouse model of Rett syndrome. Nat. Commun. 2016, 7, 10552. [Google Scholar] [CrossRef] [PubMed]
- Chapleau, C.A.; Boggio, E.M.; Calfa, G.; Percy, A.K.; Giustetto, M.; Pozzo-Miller, L. Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast. 2012, 2012, 976164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.Y.; Wu, C.; Jin, Y.; Gao, M.; Li, G.H.; Turner, D.; Shen, J.X.; Zhang, S.J.; Narayanan, V.; Jentarra, G.; et al. Electrophysiological phenotypes of MeCP2 A140V mutant mouse model. CNS Neurosci. Ther. 2014, 20, 420–428. [Google Scholar] [CrossRef]
- Stearns, N.A.; Schaevitz, L.R.; Bowling, H.; Nag, N.; Berger, U.V.; Berger-Sweeney, J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: A model for Rett syndrome. Neuroscience 2007, 146, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Levenson, J.M.; Battaglia, F.; Atkinson, R.; Teague, R.; Antalffy, B.; Armstrong, D.; Arancio, O.; Sweatt, J.D.; Zoghbi, H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef]
- Chen, P.; Liu, Z.; Zhang, Q.; Lin, D.; Song, L.; Liu, J.; Jiao, H.F.; Lai, X.; Zou, S.; Wang, S.; et al. DSCAM Deficiency Leads to Premature Spine Maturation and Autism-like Behaviors. J. Neurosci. 2022, 42, 532–551. [Google Scholar] [CrossRef]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef]
- Morello, N.; Schina, R.; Pilotto, F.; Phillips, M.; Melani, R.; Plicato, O.; Pizzorusso, T.; Pozzo-Miller, L.; Giustetto, M. Loss of Mecp2 causes atypical synaptic and molecular plasticity of parvalbumin-expressing interneurons reflecting rett syndrome–like sensorimotor defects. eNeuro 2018, 5, ENEURO.0086-18.2018. [Google Scholar] [CrossRef]
- Medrihan, L.; Tantalaki, E.; Aramuni, G.; Sargsyan, V.; Dudanova, I.; Missler, M.; Zhang, W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 2008, 99, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Asaka, Y.; Jugloff, D.G.M.; Zhang, L.; Eubanks, J.H.; Fitzsimonds, R.M. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 2006, 21, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.; Shepherd, G.M.G. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in a mutant mouse model of Rett syndrome. Neurobiol. Dis. 2010, 38, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [Green Version]
- Huo, H.Q.; Qu, Z.Y.; Yuan, F.; Ma, L.; Yao, L.; Xu, M.; Hu, Y.; Ji, J.; Bhattacharyya, A.; Zhang, S.C.; et al. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Rep. 2018, 10, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Giffin-Rao, Y.; Sheng, J.; Strand, B.; Xu, K.; Huang, L.; Medo, M.; Risgaard, K.A.; Dantinne, S.; Mohan, S.; Keshan, A.; et al. Altered patterning of trisomy 21 interneuron progenitors. Stem Cell Rep. 2022, 17, 1366–1379. [Google Scholar] [CrossRef]
- Liu, H.; Huang, S.; Wang, W.; Wang, H.; Huang, W.; Zhai, Z.; Wang, D.; Fan, Y.; Sun, J.; Li, D.; et al. Migration deficits of the neural crest caused by CXADR triplication in a human Down syndrome stem cell model. Cell Death Dis. 2022, 13, 1018. [Google Scholar] [CrossRef]
- Murray, A.; Letourneau, A.; Canzonetta, C.; Stathaki, E.; Gimelli, S.; Sloan-Bena, F.; Abrehart, R.; Goh, P.; Lim, S.; Baldo, C.; et al. Brief report: Isogenic induced pluripotent stem cell lines from an adult with mosaic down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells 2015, 33, 2077–2084. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.F.; Béna, F.; et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2014, 6, 259–277. [Google Scholar] [CrossRef]
- Xu, R.; Brawner, A.T.; Li, S.; Liu, J.J.; Kim, H.; Xue, H.; Pang, Z.P.; Kim, W.Y.; Hart, R.P.; Liu, Y.; et al. OLIG2 Drives Abnormal Neurodevelopmental Phenotypes in Human iPSC-Based Organoid and Chimeric Mouse Models of Down Syndrome. Cell Stem Cell 2019, 24, 908–926.e8. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Fogo, M.S.; Pinto, B.G.G.; Alves, A.Y.; Suzuki, A.M.; Morales, A.G.; Ezquina, S.; Sosa, O.J.; Sutton, G.J.; Sunaga-Franze, D.Y.; et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 2021, 26, 1589–1605. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xu, J.; Yang, M.Q. Gene regulation analysis reveals perturbations of autism spectrum disorder during neural system development. Genes 2021, 12, 1901. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Kasem, E.; Kurihara, T.; Tabuchi, K. Neurexins and neuropsychiatric disorders. Neurosci. Res. 2018, 127, 53–60. [Google Scholar] [CrossRef]
- Benke, T.A.; Park, K.; Krey, I.; Camp, C.R.; Song, R.; Ramsey, A.J.; Yuan, H.; Traynelis, S.F.; Lemke, J. Clinical and therapeutic significance of genetic variation in the GRIN gene family encoding NMDARs. Neuropharmacology 2021, 199, 108805. [Google Scholar] [CrossRef] [PubMed]
- Bouché, N.; Fait, A.; Zik, M.; Fromm, H. The root-specific glutamate decarboxylase (GAD1) is essential for sustaining GABA levels in Arabidopsis. Plant Mol. Biol. 2004, 55, 315–325. [Google Scholar] [CrossRef]
- Pearson, G.; Song, C.; Hohmann, S.; Prokhorova, T.; Sheldrick-Michel, T.M.; Knöpfel, T. DNA Methylation Profiles of GAD1 in Human Cerebral Organoids of Autism Indicate Disrupted Epigenetic Regulation during Early Development. Int. J. Mol. Sci. 2022, 23, 9188. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Wu, K.; Pandey, S.; Lehr, A.W.; Li, Y.; Bemben, M.A.; Badger, J.D.; Lauzon, J.L.; Wang, T.; Zaghloul, K.A.; et al. A Cluster of Autism-Associated Variants on X-Linked NLGN4X Functionally Resemble NLGN4Y. Neuron 2020, 106, 759–768.e7. [Google Scholar] [CrossRef]
- Wang, M.; Wei, P.C.; Lim, C.K.; Gallina, I.S.; Marshall, S.; Marchetto, M.C.; Alt, F.W.; Gage, F.H. Increased Neural Progenitor Proliferation in a hiPSC Model of Autism Induces Replication Stress-Associated Genome Instability. Cell Stem Cell 2020, 26, 221–233.e6. [Google Scholar] [CrossRef]
- de Jong, J.O.; Llapashtica, C.; Genestine, M.; Strauss, K.; Provenzano, F.; Sun, Y.; Zhu, H.; Cortese, G.P.; Brundu, F.; Brigatti, K.W.; et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 2021, 12, 4087. [Google Scholar] [CrossRef]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6i, 55. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef]
- Haase, F.; Gloss, B.S.; Tam, P.P.L.; Gold, W.A. Wgcna identifies translational and proteasome-ubiquitin dysfunction in rett syndrome. Int. J. Mol. Sci. 2021, 22, 9954. [Google Scholar] [CrossRef]
- Rodrigues, D.C.; Mufteev, M.; Weatheritt, R.J.; Djuric, U.; Ha, K.C.H.; Ross, P.J.; Wei, W.; Piekna, A.; Sartori, M.A.; Byres, L.; et al. Shifts in Ribosome Engagement Impact Key Gene Sets in Neurodevelopment and Ubiquitination in Rett Syndrome. Cell Rep. 2020, 30, 4179–4196.e11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.L.; Wang, Z.; Liao, Q.; Zhu, Y.; Zhou, W.H.; Xu, W.; Qiu, Z. MeCP2 Suppresses Nuclear MicroRNA Processing and Dendritic Growth by Regulating the DGCR8/Drosha Complex. Dev. Cell 2014, 28, 547–560. [Google Scholar] [CrossRef]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.N.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Varderidou-Minasian, S.; Hinz, L.; Hagemans, D.; Posthuma, D.; Altelaar, M.; Heine, V.M. Quantitative proteomic analysis of Rett iPSC-derived neuronal progenitors. Mol. Autism 2020, 11, 38. [Google Scholar] [CrossRef]
- Musi, C.A.; Castaldo, A.M.; Valsecchi, A.E.; Cimini, S.; Morello, N.; Pizzo, R.; Renieri, A.; Meloni, I.; Bonati, M.; Giustetto, M.; et al. JNK signaling provides a novel therapeutic target for Rett syndrome. BMC Biol. 2021, 19, 256. [Google Scholar] [CrossRef]
- Bassetti, D.; Lombardi, A.; Kirischuk, S.; Luhmann, H.J. Haploinsufficiency of Tsc2 leads to hyperexcitability of medial prefrontal cortex via weakening of tonic gabab receptor-mediated inhibition. Cereb. Cortex 2020, 30, 6313–6324. [Google Scholar] [CrossRef]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.M.; Hysolli, E.; Cakir, B.; Kim, K.Y.; Wang, W.; Kang, Y.J.; Clement, E.M.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98.e9. [Google Scholar] [CrossRef]
- Meharena, H.S.; Marco, A.; Dileep, V.; Lockshin, E.R.; Akatsu, G.Y.; Mullahoo, J.; Watson, L.A.; Ko, T.; Guerin, L.N.; Abdurrob, F.; et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 2022, 29, 116–130.e7. [Google Scholar] [CrossRef]
- Hirata, K.; Nambara, T.; Kawatani, K.; Nawa, N.; Yoshimatsu, H.; Kusakabe, H.; Banno, K.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; et al. 4-Phenylbutyrate ameliorates apoptotic neural cell death in Down syndrome by reducing protein aggregates. Sci. Rep. 2020, 10, 14047. [Google Scholar] [CrossRef] [PubMed]
- Sobol, M.; Klar, J.; Laan, L.; Shahsavani, M.; Schuster, J.; Annerén, G.; Konzer, A.; Mi, J.; Bergquist, J.; Nordlund, J.; et al. Transcriptome and Proteome Profiling of Neural Induced Pluripotent Stem Cells from Individuals with Down Syndrome Disclose Dynamic Dysregulations of Key Pathways and Cellular Functions. Mol. Neurobiol. 2019, 56, 7113–7127. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, P.K.; Roberts, C.M.; Fonte, V.; Jacobsen, C.; Stein, G.H.; Link, C.D. Transcriptome analysis of genetically matched human induced pluripotent stem cells disomic or trisomic for chromosome 21. PLoS ONE 2018, 13, e0194581. [Google Scholar] [CrossRef]
- El-Kordi, A.; Winkler, D.; Hammerschmidt, K.; Kästner, A.; Krueger, D.; Ronnenberg, A.; Ritter, C.; Jatho, J.; Radyushkin, K.; Bourgeron, T.; et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav. Brain Res. 2013, 251, 41–49. [Google Scholar] [CrossRef]
- Delattre, V.; La Mendola, D.; Meystre, J.; Markram, H.; Markram, K. Nlgn4 knockout induces network hypo-excitability in juvenile mouse somatosensory cortex in vitro. Sci. Rep. 2013, 3, 2897. [Google Scholar] [CrossRef]
- Unichenko, P.; Yang, J.W.; Kirischuk, S.; Kolbaev, S.; Kilb, W.; Hammer, M.; Krueger-Burg, D.; Brose, N.; Luhmann, H.J. Autism related neuroligin-4 knockout impairs intracortical processing but not sensory inputs in mouse barrel cortex. Cereb. Cortex 2018, 28, 2873–2886. [Google Scholar] [CrossRef]
- Ju, A.; Hammerschmidt, K.; Tantra, M.; Krueger, D.; Brose, N.; Ehrenreich, H. Juvenile manifestation of ultrasound communication deficits in the neuroligin-4 null mutant mouse model of autism. Behav. Brain Res. 2014, 270, 159–164. [Google Scholar] [CrossRef]
- Hammer, M.; Krueger-Burg, D.; Tuffy, L.P.; Cooper, B.H.; Taschenberger, H.; Goswami, S.P.; Ehrenreich, H.; Jonas, P.; Varoqueaux, F.; Rhee, J.S.; et al. Perturbed Hippocampal Synaptic Inhibition and γ-Oscillations in a Neuroligin-4 Knockout Mouse Model of Autism. Cell Rep. 2015, 13, 516–523. [Google Scholar] [CrossRef]
- Krueger, D.A.; Northrup, H.; Krueger, D.A.; Roberds, S.; Smith, K.; Sampson, J.; Korf, B.; Kwiatkowski, D.J.; Mowat, D.; Nellist, M.; et al. Tuberous sclerosis complex surveillance and management: Recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr. Neurol. 2013, 49, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Waltereit, R.; Japs, B.; Schneider, M.; De Vries, P.J.; Bartsch, D. Epilepsy and Tsc2 haploinsufficiency lead to autistic-like social deficit behaviors in rats. Behav. Genet. 2011, 41, 364–372. [Google Scholar] [CrossRef]
- Neklyudova, A.; Smirnov, K.; Rebreikina, A.; Martynova, O.; Sysoeva, O. Electrophysiological and Behavioral Evidence for Hyper-and Hyposensitivity in Rare Genetic Syndromes Associated with Autism. Genes 2022, 13, 671. [Google Scholar] [CrossRef]
- Sundberg, M.; Tochitsky, I.; Buchholz, D.E.; Winden, K.; Kujala, V.; Kapur, K.; Cataltepe, D.; Turner, D.; Han, M.J.; Woolf, C.J.; et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry 2018, 23, 2167–2183. [Google Scholar] [CrossRef]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Wundrach, D.; Martinetti, L.E.; Stafford, A.M.; Bilinovich, S.M.; Angara, K.; Prokop, J.W.; Crandall, S.R.; Vogt, D. A Human TSC1 Variant Screening Platform in Gabaergic Cortical Interneurons for Genotype to Phenotype Assessments. Front. Mol. Neurosci. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Moore, S.M.; Seidman, J.S.; Ellegood, J.; Gao, R.; Savchenko, A.; Troutman, T.D.; Abe, Y.; Stender, J.; Lee, D.; Wang, S.; et al. Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl. Psychiatry 2019, 9, 24. [Google Scholar] [CrossRef]
- Deliu, E.; Arecco, N.; Morandell, J.; Dotter, C.P.; Contreras, X.; Girardot, C.; Käsper, E.L.; Kozlova, A.; Kishi, K.; Chiaradia, I.; et al. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 2018, 21, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Hysolli, E.; Park, I.H. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14169–14174. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef]
- Cheung, A.Y.L.; Horvath, L.M.; Grafodatskaya, D.; Pasceri, P.; Weksberg, R.; Hotta, A.; Carrel, L.; Ellis, J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011, 20, 2103–2115. [Google Scholar] [CrossRef]
- Chen, X.; Han, X.; Blanchi, B.; Guan, W.; Ge, W.; Yu, Y.C.; Sun, Y.E. Graded and pan-neural disease phenotypes of Rett Syndrome linked with dosage of functional MeCP2. Protein Cell 2021, 12, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.W.M.; Marcy, G.; Yoon, S.I.; Ma, D.; Rosales, F.J.; Augustine, G.J.; Goh, E.L.K. Choline Ameliorates Disease Phenotypes in Human iPSC Models of Rett Syndrome. NeuroMolecular Med. 2016, 18, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Ananiev, G.; Williams, E.C.; Li, H.; Chang, Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from rett syndrome patients as In Vitro disease model. PLoS ONE 2011, 6, e25255. [Google Scholar] [CrossRef]
- Djuric, U.; Cheung, A.Y.L.; Zhang, W.; Mok, R.S.; Lai, W.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.S.; et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB signaling is involved in rett syndrome pathogenesis. J. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Silva-Fernandes, A.; Oliveira, P.; Sousa, N.; Maciel, P. Evidence for abnormal early development in a mouse model of Rett syndrome. Genes, Brain Behav. 2007, 6, 277–286. [Google Scholar] [CrossRef]
- Rietveld, L.; Stuss, D.P.; McPhee, D.; Delaney, K.R. Genotype-specific effects of Mecp2 loss-of-function on morphology of layer V pyramidal neurons in heterozygous female Rett syndrome model mice. Front. Cell. Neurosci. 2015, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Itoh, M.; Ichikawa, T.; Washiyama, K.; Goto, Y.I. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 2005, 64, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, J.V.; Martínez-Rodríguez, E.; Forte, A.; Blanco-Gómez, C.; Stork, O.; Lanuza, E.; Santos, M.; Agustín-Pavón, C. Early life stress exacerbates behavioural and neuronal alterations in adolescent male mice lacking methyl-CpG binding protein 2 (Mecp2). Front. Behav. Neurosci. 2022, 16, 974692. [Google Scholar] [CrossRef]
- Fidler, D.J.; Prince, M.A.; Van Deusen, K.; Esbensen, A.J.; Thurman, A.J.; Abbeduto, L.; Patel, L.; Mervis, C.; Schworer, E.K.; Lee, N.R.; et al. Latent profiles of autism symptoms in children and adolescents with Down syndrome. J. Intellect. Disabil. Res. 2022, 66, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.C.; Capone, G.T.; Kaufmann, W.E. Neuroanatomic correlates of autism and stereotypy in children with Down syndrome. Neuroreport 2008, 19, 653–656. [Google Scholar] [CrossRef]
- Larsen, K.B.; Laursen, H.; Græm, N.; Samuelsen, G.B.; Bogdanovic, N.; Pakkenberg, B. Reduced cell number in the neocortical part of the human fetal brain in Down syndrome. Ann. Anat. 2008, 190, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Utagawa, E.C.; Moreno, D.G.; Schafernak, K.T.; Arva, N.C.; Malek-Ahmadi, M.H.; Mufson, E.J.; Perez, S.E. Neurogenesis and neuronal differentiation in the postnatal frontal cortex in Down syndrome. Acta Neuropathol. Commun. 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Kesslak, J.P.; Nagata, S.F.; Lott, I.; Nalcioglu, O. Magnetic resonance imaging analysis of age–related changes in the brains of individuals with down’s syndrome. Neurology 1994, 44, 1039–1045. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Almeida, O.F.X.; Takashima, A. Local Somatodendritic Translation and Hyperphosphorylation of Tau Protein Triggered by AMPA and NMDA Receptor Stimulation. EBioMedicine 2017, 20, 120–126. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Uguagliati, B.; Stagni, F.; Emili, M.; Giacomini, A.; Russo, C.; Guidi, S.; Bartesaghi, R. Early Appearance of Dendritic Alterations in Neocortical Pyramidal Neurons of the Ts65Dn Model of Down Syndrome. Dev. Neurosci. 2022, 44, 23–38. [Google Scholar] [CrossRef]
- Alldred, M.J.; Lee, S.H.; Petkova, E.; Ginsberg, S.D. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-middle-aged Ts65Dn mice. J. Comp. Neurol. 2015, 523, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, L.; Best, T.K.; Cramer, N.P.; Carney, R.S.E.; Isaac, J.T.R.; Galdzicki, Z.; Haydar, T.F. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 2010, 13, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Kleschevnikov, A.M.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009, 512, 453–466. [Google Scholar] [CrossRef]
- Pérez-Cremades, D.; Hernández, S.; Blasco-Ibáñez, J.M.; Crespo, C.; Nacher, J.; Varea, E. Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down’s syndrome. J. Neural Transm. 2010, 117, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Weick, J.P.; Held, D.L.; Bonadurer, G.F.; Doers, M.E.; Liu, Y.; Maguire, C.; Clark, A.; Knackert, J.A.; Molinarolo, K.; Musser, M.; et al. Deficits in human trisomy 21 iPSCs and neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 9962–9967. [Google Scholar] [CrossRef]
- Mizuno, G.O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant Calcium Signaling in Astrocytes Inhibits Neuronal Excitability in a Human Down Syndrome Stem Cell Model. Cell Rep. 2018, 24, 355–365. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; McMillan, E.; Chen, S.I.; Wallace, K.; Svendsen, C.N. A critical period in cortical interneuron neurogenesis in down syndrome revealed by human neural progenitor cells. Dev. Neurosci. 2009, 31, 497–510. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Buttterfield, D.A. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: Insights from proteomics. Proteom.-Clin. Appl. 2014, 8, 73–85. [Google Scholar] [CrossRef]
- Perluigi, M.; di Domenico, F.; Fiorini, A.; Cocciolo, A.; Giorgi, A.; Foppoli, C.; Butterfield, D.A.; Giorlandino, M.; Giorlandino, C.; Eugenia Schininà, M.; et al. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteom.-Clin. Appl. 2011, 5, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Corrales, A.; Parisotto, E.B.; Vidal, V.; García-Cerro, S.; Lantigua, S.; Diego, M.; Wilhem Filho, D.; Sanchez-Barceló, E.J.; Martínez-Cué, C.; Rueda, N. Pre- and post-natal melatonin administration partially regulates brain oxidative stress but does not improve cognitive or histological alterations in the Ts65Dn mouse model of Down syndrome. Behav. Brain Res. 2017, 334, 142–154. [Google Scholar] [CrossRef]
- Tang, X.Y.; Xu, L.; Wang, J.; Hong, Y.; Wang, Y.; Zhu, Q.; Wang, D.; Zhang, X.Y.; Liu, C.Y.; Fang, K.H.; et al. DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with down syndrome. J. Clin. Investig. 2021, 131, e135763. [Google Scholar] [CrossRef]
- Qiu, J.J.; Liu, Y.N.; Ren, Z.R.; Yan, J.B. Dysfunctions of mitochondria in close association with strong perturbation of long noncoding RNAs expression in down syndrome. Int. J. Biochem. Cell Biol. 2017, 92, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Mollo, N.; Esposito, M.; Aurilia, M.; Scognamiglio, R.; Accarino, R.; Bonfiglio, F.; Cicatiello, R.; Charalambous, M.; Procaccini, C.; Micillo, T.; et al. Human trisomic ipscs from down syndrome fibroblasts manifest mitochondrial alterations early during neuronal differentiation. Biology 2021, 10, 609. [Google Scholar] [CrossRef]
- Xu, L.; Huo, H.Q.; Lu, K.Q.; Tang, X.Y.; Hong, Y.; Han, X.; Fu, Z.X.; Fang, K.H.; Xu, M.; Guo, X.; et al. Abnormal mitochondria in Down syndrome iPSC-derived GABAergic interneurons and organoids. Biochim. Biophys. Acta-Mol. Basis Dis. 2022, 1868, 166388. [Google Scholar] [CrossRef] [PubMed]
- Laan, L.; Klar, J.; Sobol, M.; Hoeber, J.; Shahsavani, M.; Kele, M.; Fatima, A.; Zakaria, M.; Annerén, G.; Falk, A.; et al. DNA methylation changes in down syndrome derived neural iPSCs uncover co-dysregulation of ZNF and HOX3 families of transcription factors. Clin. Epigenet. 2020, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Casañas, J.J.; González-Corrales, M.; Urbano-Gámez, J.D.; Alves-Sampaio, A.; Troca-Marín, J.A.; Montesinos, M.L. CPEB1 is overexpressed in neurons derived from Down syndrome IPSCs and in the hippocampus of the mouse model Ts1Cje. Mol. Cell. Neurosci. 2019, 95, 79–85. [Google Scholar] [CrossRef]
- Wu, C.I.; Vinton, E.A.; Pearse, R.V.; Heo, K.; Aylward, A.J.; Hsieh, Y.C.; Bi, Y.; Adeleye, S.; Fancher, S.; Duong, D.M.; et al. APP and DYRK1A regulate axonal and synaptic vesicle protein networks and mediate Alzheimer’s pathology in trisomy 21 neurons. Mol. Psychiatry 2022, 27, 1970–1989. [Google Scholar] [CrossRef]
- Czermiński, J.T.; Lawrence, J.B. Silencing Trisomy 21 with XIST in Neural Stem Cells Promotes Neuronal Differentiation. Dev. Cell 2020, 52, 294–308.e3. [Google Scholar] [CrossRef]
- Paşca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Paşca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Freed, W.J.; Mash, D.C. CNVs in neurodevelopmental disorders. Oncotarget 2015, 6, 18238–18239. [Google Scholar] [CrossRef]
- Soeda, S.; Saito, R.; Fujita, N.; Fukuta, K.; Taniura, H. Neuronal differentiation defects in induced pluripotent stem cells derived from a Prader-Willi syndrome patient. Neurosci. Lett. 2019, 703, 162–167. [Google Scholar] [CrossRef]
- Soeda, S.; Saito, R.; Fujii, A.; Tojo, S.; Tokumura, Y.; Taniura, H. Abnormal DNA methylation in pluripotent stem cells from a patient with Prader-Willi syndrome results in neuronal differentiation defects. Stem Cell Res. 2021, 53, 102351. [Google Scholar] [CrossRef]
- Fink, J.J.; Robinson, T.M.; Germain, N.D.; Sirois, C.L.; Bolduc, K.A.; Ward, A.J.; Rigo, F.; Chamberlain, S.J.; Levine, E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017, 8, 15038. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.J.; Schreiner, J.D.; Bloom, J.E.; James, J.; Baker, D.S.; Robinson, T.M.; Lieberman, R.; Loew, L.M.; Chamberlain, S.J.; Levine, E.S. Hyperexcitable Phenotypes in Induced Pluripotent Stem Cell–Derived Neurons From Patients With 15q11-q13 Duplication Syndrome, a Genetic Form of Autism. Biol. Psychiatry 2021, 90, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Yadav, S.; Dao, D.Q.; Wu, Z.Y.; Hokanson, K.C.; Cahill, M.K.; Wiita, A.P.; Jan, Y.N.; Ullian, E.M.; Weiss, L.A. Cellular Phenotypes in Human iPSC-Derived Neurons from a Genetic Model of Autism Spectrum Disorder. Cell Rep. 2017, 21, 2678–2687. [Google Scholar] [CrossRef] [PubMed]
- Klingler, F.M.; Gastreich, M.; Grygorenko, O.O.; Savych, O.; Borysko, P.; Griniukova, A.; Gubina, K.E.; Lemmen, C.; Moroz, Y.S. SAR by space: Enriching hit sets from the chemical space. Molecules 2019, 24, 3096. [Google Scholar] [CrossRef]
- Odawara, A.; Matsuda, N.; Ishibashi, Y.; Yokoi, R.; Suzuki, I. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Sci. Rep. 2018, 8, 10416. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.; Parham, F.; Dea, S.; Sodhi, N.; Biesmans, S.; Mora-Castilla, S.; Ryan, K.; Behl, M.; Chandy, G.; Crittenden, C.; et al. Functional and mechanistic neurotoxicity profiling using human iPSC-Derived neural 3D cultures. Toxicol. Sci. 2019, 167, 249–257. [Google Scholar] [CrossRef]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. eBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef]
- Dickstein, D.P.; Towbin, K.E.; Van Der Veen, J.W.; Rich, B.A.; Brotman, M.A.; Knopf, L.; Onelio, L.; Pine, D.S.; Leibenluft, E. Randomized double-blind placebo-controlled trial of lithium in youths with severe mood dysregulation. J. Child Adolesc. Psychopharmacol. 2009, 19, 61–73. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, H.M.; Kaufmann, W.E.; Barnes, K.V.; Rakesh, K.; Kapur, K.; Tarquinio, D.C.; Cantwell, N.G.; Roche, K.J.; Rose, S.A.; Walco, A.C.; et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann. Clin. Transl. Neurol. 2018, 5, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Devesa, O.; Carrillo, M.; Casteleiro, N.; Devesa, A.; Llorente, D.; González, C. Rett Syndrome: Treatment with IGF-I, Melatonin, Blackcurrant Extracts, and Rehabilitation. Reports 2018, 1, 14. [Google Scholar] [CrossRef]
- Linker, S.B.; Mendes, A.P.D.; Marchetto, M.C.; Marchetto, M.C. IGF-1 treatment causes unique transcriptional response in neurons from individuals with idiopathic autism. Mol. Autism 2020, 11, 55. [Google Scholar] [CrossRef]
- Glaze, D.G.; Neul, J.L.; Kaufmann, W.E.; Berry-Kravis, E.; Condon, S.; Stoms, G.; Oosterholt, S.; Della Pasqua, O.; Glass, L.; Jones, N.E.; et al. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology 2019, 92, E1912–E1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neul, J.L.; Percy, A.K.; Benke, T.A.; Berry-Kravis, E.M.; Glaze, D.G.; Peters, S.U.; Jones, N.E.; Youakim, J.M. Design and outcome measures of LAVENDER, a phase 3 study of trofinetide for Rett syndrome. Contemp. Clin. Trials 2022, 114, 106704. [Google Scholar] [CrossRef]
- Croci, S.; Carriero, M.L.; Capitani, K.; Daga, S.; Donati, F.; Papa, F.T.; Frullanti, E.; Lopergolo, D.; Lamacchia, V.; Tita, R.; et al. AAV-mediated FOXG1 gene editing in human Rett primary cells. Eur. J. Hum. Genet. 2020, 28, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, K.K.E.; Vudhironarit, T.; Hector, R.D.; Sinnett, S.; Bahey, N.G.; Bailey, M.E.S.; Gray, S.J.; Cobb, S.R. Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol. Ther. Methods Clin. Dev. 2017, 5, 180–190. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP2 -KO neurons and cortical organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
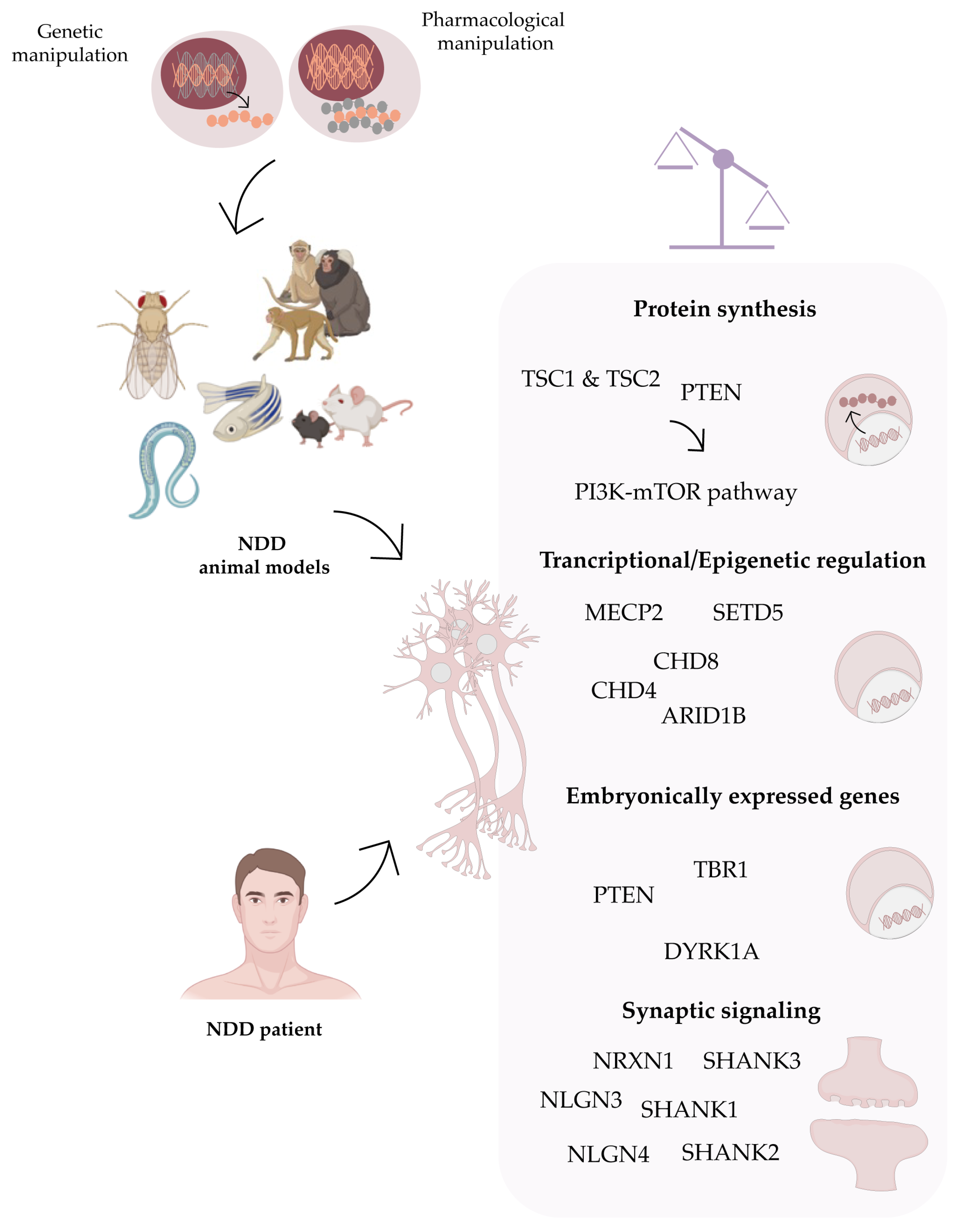


| Animal Model | Animal | Advantages | Disadvantages | References |
|---|---|---|---|---|
Drosophila melanogaster | - | Easy to measure behavioral phenotypes | [22,23,24] | |
| Reduced size | ||||
| Easy to manipulate genetically | ||||
| Absence of genetic redundancy | Lack of neuronal complexity | |||
| Cost-effective | Organ and physiological disparity from humans | |||
| Easy maintenance | Lower translatability to clinics | |||
| Short-life cycle | ||||
| Suitable for large-scale drug and genetic screenings | ||||
Caenorhabditis elegans | - | Well-characterized nervous system | [25,26] | |
| Transparent body | ||||
| Reduced size | ||||
| Easy to manipulate genetically | Lack of neuronal complexity | |||
| Cost-efficient | Organ and physiological disparity from humans | |||
| Easy maintenance | Lower translatability to clinics | |||
| Short-life cycle | ||||
| Suitable for large-scale drug and genetic screenings | ||||
Zebrafish (Danio rerio) | - | Short reproductive cycle | [27,28] | |
| Presence of a brain | ||||
| Transparent embryos and larvae | Lack of neuronal complexity | |||
| Cost-efficient | Lower translatability to clinics | |||
| Useful for drug and genetic screenings | ||||
| Presents well-defined circuits with conserved synaptic systems | ||||
Rodent models | Mice (Mus musculus) Rats (Rattus norvegicus) | High face validity | [29,30,31] | |
| High resemblance to humans | ||||
| Similar molecular pathways affected in disease | Insufficient cortical and circuitry complexity | |||
| Some cognitive and social behaviors resembling humans’ | ||||
| Amenable to genetic manipulation | ||||
| Suitable for pre-clinical drug testing | ||||
Non-human primates | Cynomolgus | Highest resemblance to human genetics, brain structure and cognitive function | Demanding genomic editing strategies | [32,33] |
| Rhesus macaque | Higher cognitive and social behaviors resemblance to humans | High cost | ||
| Marmoset | Amenable to genetic manipulation | Long developmental time | ||
| Suitable for pre-clinical drug testing | ||||
 | ||||
| NDD | Variant | Human | Rodents | hIPSCs |
|---|---|---|---|---|
| ASD | Small stature | - | ||
| Smaller cerebellum | ||||
| Larger hippocampus | ||||
| Altered vocalizations | ||||
| Corpus callosum underdevelopment/absence | Social, learning and memory impairments | |||
| Characteristic facial features | Anxiety-like behavior | |||
| ARID1B deletion [308,309,310,311,312,313,314] | Microcephaly | Cortical E/I imbalance | ||
| Cognitive deficits | Decreased excitatory synaptic density and transmission | |||
| ID | Decreased number of cortical GABAergic interneurons | |||
| Reduced proliferation of interneuron NPCs | ||||
| PV+ and SST+ interneurons impairments | ||||
| CHD4 mutant (r.Arg11276Gln, p.Trp1148Leu, p.Arg1173Leu, p.Gly1003Asp) [315,316] | Developmental delay, facial dysmorphisms, macrocephaly, ID | - | - | |
| Macrocephaly | ||||
| Craniofacial abnormalities | ||||
| Increased anxiety | ||||
| Repetitive behavior | ||||
| ASD | ||||
| Reduced E/I balance | ||||
| Fast early postnatal growth | CHD8 predominantly expressed in MAP2+ and PV+ neurons | |||
| Macrocephaly | Reduced axon and dendritic growth | Altered expression of genes related with brain volume | ||
| CHD8 deletion [317,318,319,320,321,322,323,324,325,326,327,328] | Abnormal facial features | Axonal projections disruption | ||
| CHD8 is predominantly expressed in MAP2+ and PV+ neurons | Delayed neuronal migration | |||
| Altered synaptic physiology in medium spiny neurons | ||||
| Increased synchronized activity in cortico-hippocampal and auditory–parietal networks | ||||
| Increased neuronal proliferation | ||||
| Impaired social interaction and memory | ||||
| Stereotypic behaviors | Decreased NMDAR-mediated currents | |||
| DSCAM deletion [302,329] | ASD | Premature spine maturation | Downregulation of NMDAR subunits | |
| Excessive glutamatergic transmission | ||||
| Reduced NMDAR currents | ||||
| ASD | Decreased growth cone size Increased soma size | |||
| Developmental delay | Reduced social interaction and vocalizations | Increased neuronal proliferation | ||
| SHANK2 deletion [297,298,330] | ASD | Stereotypic behavior | Decreased apoptosis | |
| ID | Altered spine volume | Increased dendritic length, dendrite complexity, synapse number and frequency of sEPSCs | ||
| Decreased NMDAR function | ||||
| Juvenile impaired social interaction | ||||
| Enhanced self-grooming | ||||
| Anxiety-like behavior | ||||
| Social dominance behavior | ||||
| Motor abnormalities | ||||
| SHANK3 mutant (R11117X, Q321R, c.1527G > A, c.2497delG, C.5008A > T, ctMUT, RS9616915SNP, exon (e)4-9, e14-16, S685) [331,332,333,334,335,336,337,338,339,340,341,342,343,344,345] | Abnormal development of sleep and arousal mechanisms | Altered spinogenesis of pyramidal cortical neurons | ||
| Reduced cell soma size | ||||
| Striatal synaptic transmission defects, before winning | ||||
| PFC synaptic defects | ||||
| Decreased neuronal excitability | ||||
| Deficient LTP | ||||
| Developmental delay | Reduced complexity of dendritic tree, spines and excitatory synapses | |||
| Language delay | Repetitive grooming | |||
| Social deficits | ||||
| ASD | Anxiety-like behavior | |||
| Motor deficits | ||||
| Altered SHANK3 methylation pattern | Altered light sensitivity | |||
| Reduced corpus callosum volume | Impaired neuronal development | |||
| NPCs early differentiation | Impaired mature neuronal function | |||
| SHANK3 deletion [346,347,348,349,350,351,352,353,354,355,356,357,358,359,360] | Increased cortical pyramidal neurons firing | Reduced neuronal soma size, growth cone area, neurite length and branch numbers | ||
| Abnormal striatal circuitry development | Defects in E/I synaptic transmission | |||
| Reduced social memory—CA1 neurons | ||||
| Glutamatergic but not GABAergic activity altered in CA3 at birth | ||||
| Decreased cortical interneurons activity | ||||
| Reduced cortical PV+ mRNA and protein levels | ||||
| Decreased PV+ basket cells in the somatosensory cortex | ||||
| ASD | Immature neurons | |||
| Over-grooming | Altered differentiation | |||
| Developmental delay | Altered social response | Reduced neuronal activity | ||
| Facial abnormalities | Cognitive deficits | Reduced number of APs | ||
| NRXN1 deletion [75,77,304,361,362,363,364,365,366,367,368] | ASD | Aggressive behavior in males | Decreased neurite number | |
| Severe breathing problems | Sex-dependent altered novelty response | Decreased neuronal length | ||
| Decrease synaptic strength in spiny projection neurons pathway | Increased cortical calcium signaling, sodium currents, AP amplitude | |||
| Reduction in neurotransmitter release in spiny neurons | Increased depolarization time | |||
| ASD | Sociability deficits | Decreased E/I ration | ||
| Reduced ultrasonic vocalization | Decreased E/I network response | |||
| NLGN4 deletion [67,369,370,371,372,373] | Increased stereotypies | Decreased miniature EPSCs and IPSCs | ||
| Cognitive dysfunction | Decreased number of GABAergic and glutamatergic vesicles | |||
| Altered GABAergic hippocampal function | Impaired synaptic formation in NPCs | |||
| TSC1/TSC2 loss of function [374] | - | - | ||
| Microcephaly | Impaired memory and UV vocalization | |||
| Short stature Abnormal facial features | Social deficits | Increased cell size, NPC proliferation and neurite outgrowth | ||
| ID | Seizures | Impaired neuronal differentiation | ||
| Seizures | Increased cell size | E/I ratio imbalance | ||
| TSC2 deletion [304,361,375,376,377,378] | Memory deficits | Increased PV levels | Neuronal hyperactivity | |
| Hypoconnected neural networks | At P7, Pax2+ cells increased and delayed maturation into PV+ interneurons | Neuronal network dysfunction | ||
| Decreased AP after hyperpolarization | Reduced synchronization of neuronal bursting and spatial connectivity | |||
| Abnormal LTP | Decreased expression of synaptic markers | |||
| ASD | Hyperactivity, social and cognitive deficits | |||
| TSC1 mutant (R336W, T360N, T393I, S403L and H732Y) [379] | Altered brain anatomy | - | ||
| Reduced cortical thickness | ||||
| Reduced cortical synaptic density and neurite outgrowth | - | |||
| SETD5 deletion [380,381] | ASD | Decreased network activity and synchrony | ||
| Enhanced LTP | - | |||
| Abnormal expression of postsynaptic density proteins |
| NDD | Variant | Human | Rodents | hIPSCs |
|---|---|---|---|---|
| Rett Syndrome | Decreased Ca2+ signaling | |||
| Decreased sEPSCs and sIPSCs | ||||
| Impaired spatial, contextual fear and social memory | Decreased synaptic contacts | |||
| Anxiety | Decreased cell soma size, dendritic branching, spine density | |||
| Increased vocalizations | Impaired neuronal maturation | |||
| Microcephaly | Hypoactivity | Increased VIP+ and CB2+ interneurons | ||
| Impaired development | Stereotypies | Decreased PV+, SST+ and CB1+ interneurons | ||
| Mild ID | Reduced brain volume | Loss of low frequency and gamma oscillations | ||
| MECP2 mutant (R168X, p24hospho(Thr308-Ser421), tm1.1Jae, tm1.1Bird, A140V, 1lox, 308) [323,324,325,326,327,328,390] | Seizures | Seizures | Decreased density of excitatory puncta | |
| Stereotypic behavior | Motor abnormalities | Premature development of deep-cortical layer | ||
| Anxiety | Breathing abnormalities | Expression of progenitor and proliferative cells | ||
| Breathing problems | Impaired growth maturation | Defective forebrain neuronal function | ||
| Motor abnormalities | Reduced cortical spontaneous activity of pyramidal neurons | Interneurons migration deficits | ||
| Reduced miniature EPSCs amplitude, without changes in miniature IPSCs | Decreased Nkx2.1 levels | |||
| Lower dendritic spine density in CA1 neurons, at P7, abolished at P15 | Increased input resistance | |||
| Deficits in LTP and LTD | Impaired voltage-gated Na+/K+ currents | |||
| Decreased dendritic complexity | ||||
| Gait and posture abnormalities | ||||
| Breathing difficulties | ||||
| Microcephaly | ||||
| Seizures | Decreased soma area, dendritic length | |||
| Stereotypic behaviors | Decreased neurite outgrowth | |||
| ID | Hypoactivity | Reduced depolarized resting membrane potential | ||
| ASD | E/I imbalance | Reduced cell capacitance | ||
| MECP2 deletion [331,332,333,334,335,391,392,393] | Epilepsy | Reduced excitatory networks | Deficits in excitatory synapse transmission | |
| Motor abnormalities | Reduced cortical basal dendritic length | Decreased excitatory markers | ||
| Delayed cortical neuronal maturation | Decreased neuronal migration | |||
| Reduced cortical neurons soma and nuclei size | Network dysfunction caused by GE-derived interneurons malfunction | |||
| Premature synaptogenesis | Increased inhibitory synapses and markers | |||
| Elevated cortical PV expression | ||||
| Abnormal excitatory inputs converging onto PV+ interneurons | ||||
| FOXG1 mutant [322] | Altered craniofacial structure | Altered craniofacial structure | - |
| NDD | Variant | Human | Rodents | hIPSCs |
|---|---|---|---|---|
| Down Syndrome | Short stature | |||
| Decreased brain size | Reduced brain organoid size | |||
| Decreased white matter volume | Reduced GABAergic interneurons | |||
| Hippocampal and neocortical size | Cognitive deficits (learning, memory and executive function) | Reduced CR+/CB+ interneurons ratio | ||
| Characteristic face phenotype Alzheimer’s disease-like histopathology | Increased number of inhibitory neurons in the forebrain | Reduced COUP-TFII+ progenitors and proliferation | ||
| ID | Dendritic spine defects | Increased OLIG2+ ventral forebrain progenitors | ||
| Cognitive deficits (learning, memory and executive function) | Decreased GABA-mediated inhibition | Reduced interneuron lineage-determining transcription factors | ||
| Chromosome 21 trisomy [336,341,394,395,396,397,398,399,400,401,402,403,404,405,406,407,408,409,410,411] | Decreased excitatory and inhibitory neurons | Decreased GABA synthesis enzymes in hippocampal and cortical inhibitory synapses | Increased GABAergic interneurons production | |
| Decreased number of forebrain neurons | Altered dendritic length | Decreased GABAergic interneuron neurogenesis and proliferation | ||
| Decreased CB+ and PV+ neurons | Altered CA1 pyramidal neuron function | Decreased synaptic activity | ||
| Decreased CB+ and PV+ size neurons | Decreased LTP | Decreased migration capacity | ||
| Dendritic spine defects | Decreased neurogenesis and proliferation | Abnormal neuronal differentiation dependent on DYRK1A | ||
| Altered neuronal migration and differentiation | Oxidative stress | Decreased mitochondrial membrane potential | ||
| Oxidative stress | Increased mitochondria number | |||
| Decreased neurogenesis and proliferation | Abnormal mitochondria morphology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerreiro, S.; Maciel, P. Transition from Animal-Based to Human Induced Pluripotent Stem Cells (iPSCs)-Based Models of Neurodevelopmental Disorders: Opportunities and Challenges. Cells 2023, 12, 538. https://doi.org/10.3390/cells12040538
Guerreiro S, Maciel P. Transition from Animal-Based to Human Induced Pluripotent Stem Cells (iPSCs)-Based Models of Neurodevelopmental Disorders: Opportunities and Challenges. Cells. 2023; 12(4):538. https://doi.org/10.3390/cells12040538
Chicago/Turabian StyleGuerreiro, Sara, and Patrícia Maciel. 2023. "Transition from Animal-Based to Human Induced Pluripotent Stem Cells (iPSCs)-Based Models of Neurodevelopmental Disorders: Opportunities and Challenges" Cells 12, no. 4: 538. https://doi.org/10.3390/cells12040538
APA StyleGuerreiro, S., & Maciel, P. (2023). Transition from Animal-Based to Human Induced Pluripotent Stem Cells (iPSCs)-Based Models of Neurodevelopmental Disorders: Opportunities and Challenges. Cells, 12(4), 538. https://doi.org/10.3390/cells12040538