

Dual Targeting of EGFR and MTOR Pathways Inhibits Glioblastoma Growth by Modulating the Tumor Microenvironment

 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Generation of GBM PDXs and the U937 Cell Line
2.2. High-Throughput Drug Screening
2.2.1. Set-Up for Pharmacological Screens
2.2.2. Quantification of Drug Response
2.3. Phospho-Kinase Array
2.4. Western Blot
2.5. Apoptosis Assays
2.6. In Vivo GBM Model and Treatment Study
2.7. RNA Analysis Using Microarray
2.8. Taqman Analysis
2.9. Immunohistochemistry
2.10. ELISA for Human Periostin and CCL2
2.11. Transwell Migration Assays
3. Results
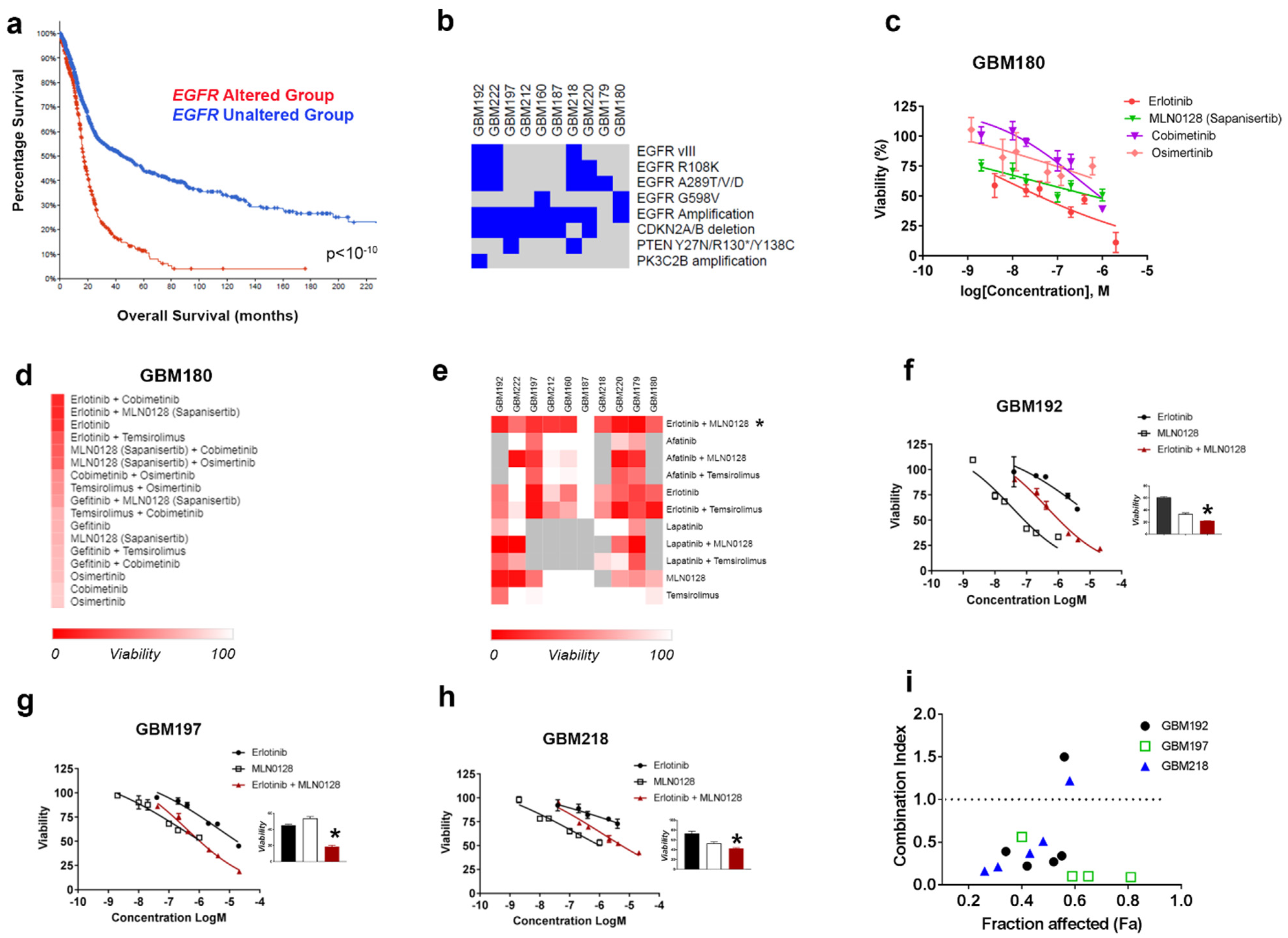
3.1. HTDS Identify the Combination of Erlotinib and MLN0128 as Most Effective and Synergistically Inhibiting the Growth of EGFR-Driven GBMs
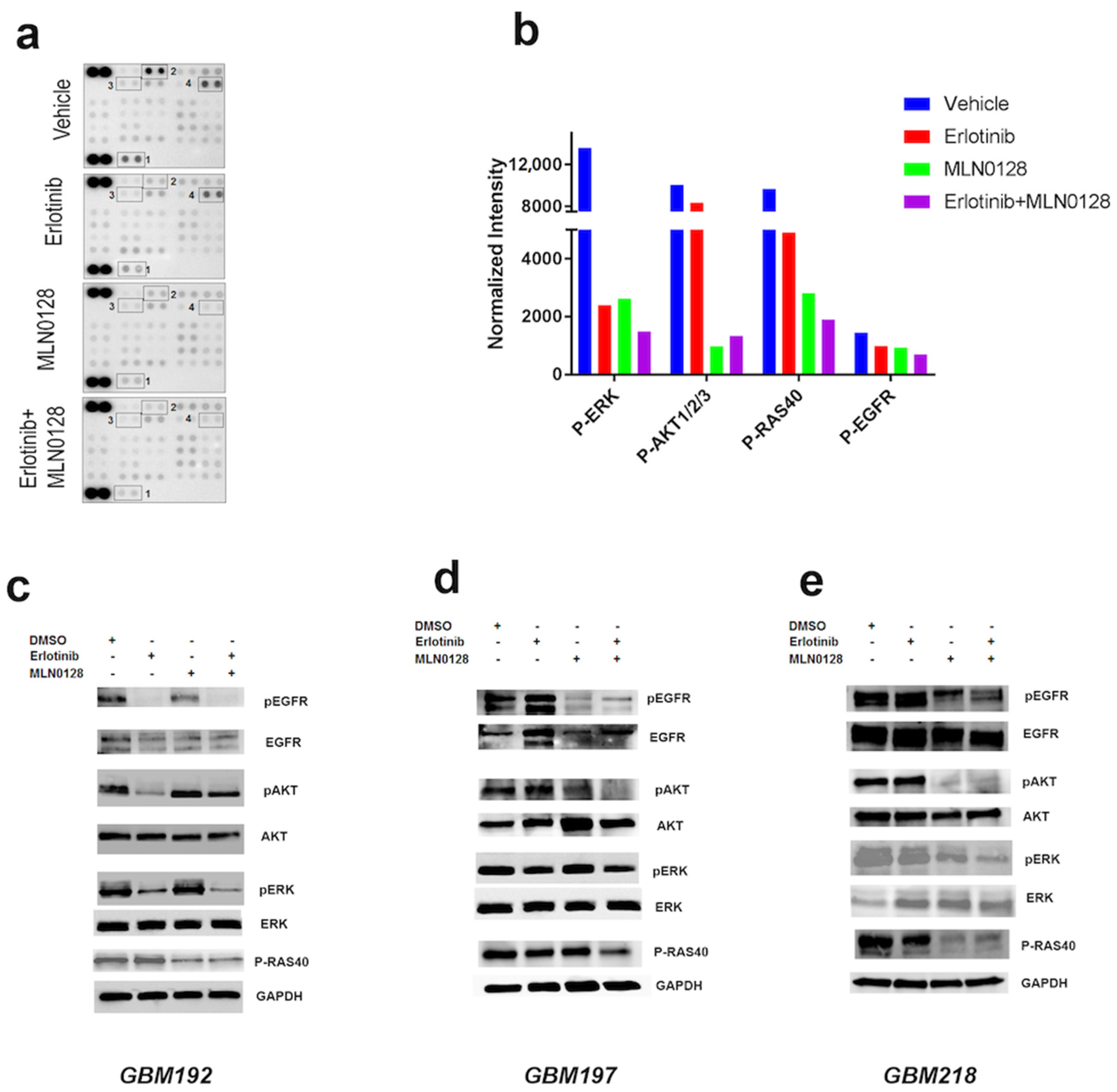
3.2. Erlotinib and MLN0128 Impact EGFR-Driven GBM Cell Survival by Inhibiting the Tumor-Promoting Pathways p-EGFR, MAPK, and PI3K/AKT/MTOR and Inducing Apoptosis
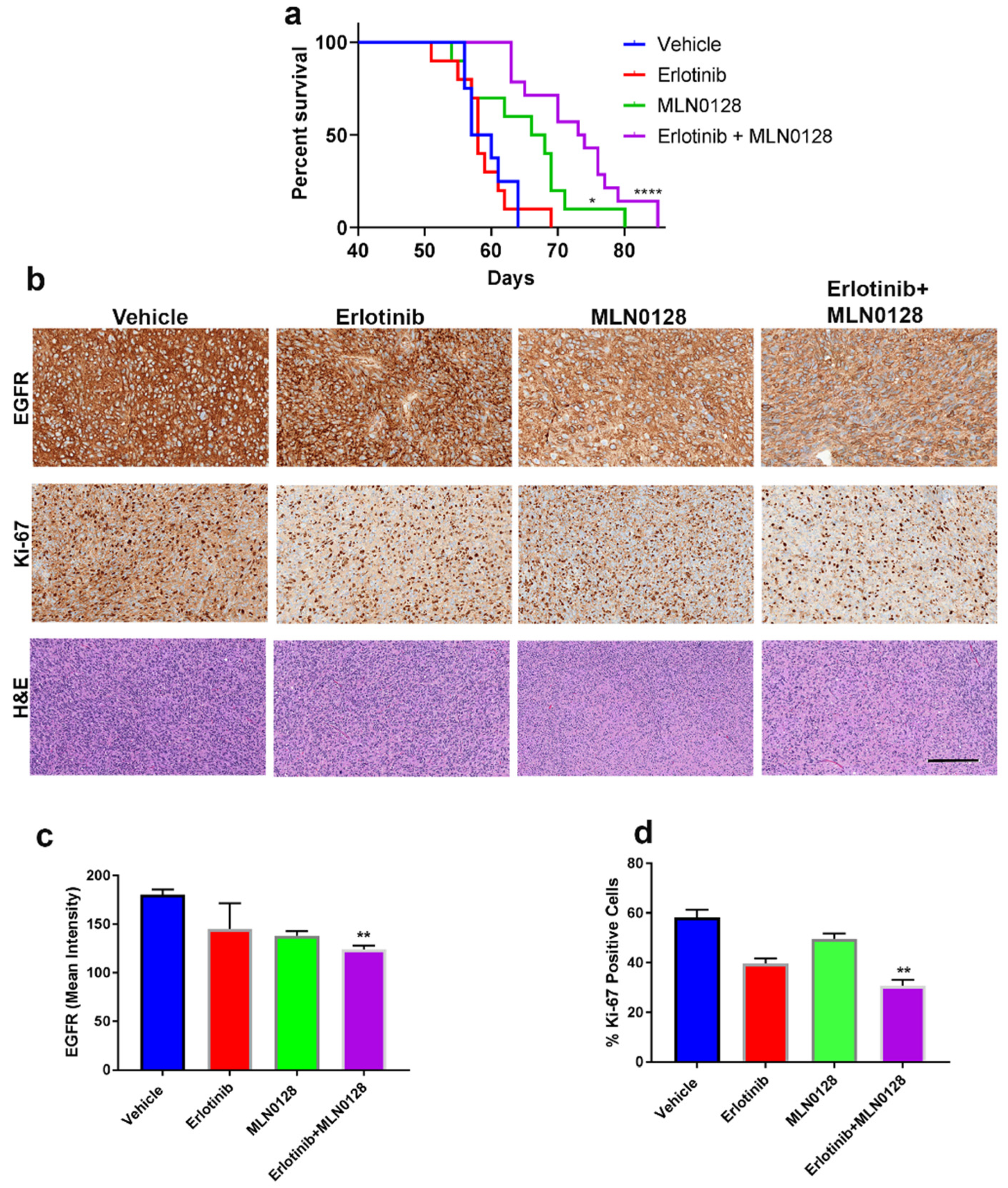
3.3. The Combination of Erlotinib and MLN0128 Prolongs the Survival of GBM-Bearing Mice
3.4. Transcriptomic Analyses of GBM Treated In Vivo Reveal Modulation of the Tumor Immune Microenvironment by Erlotinib and MLN0128
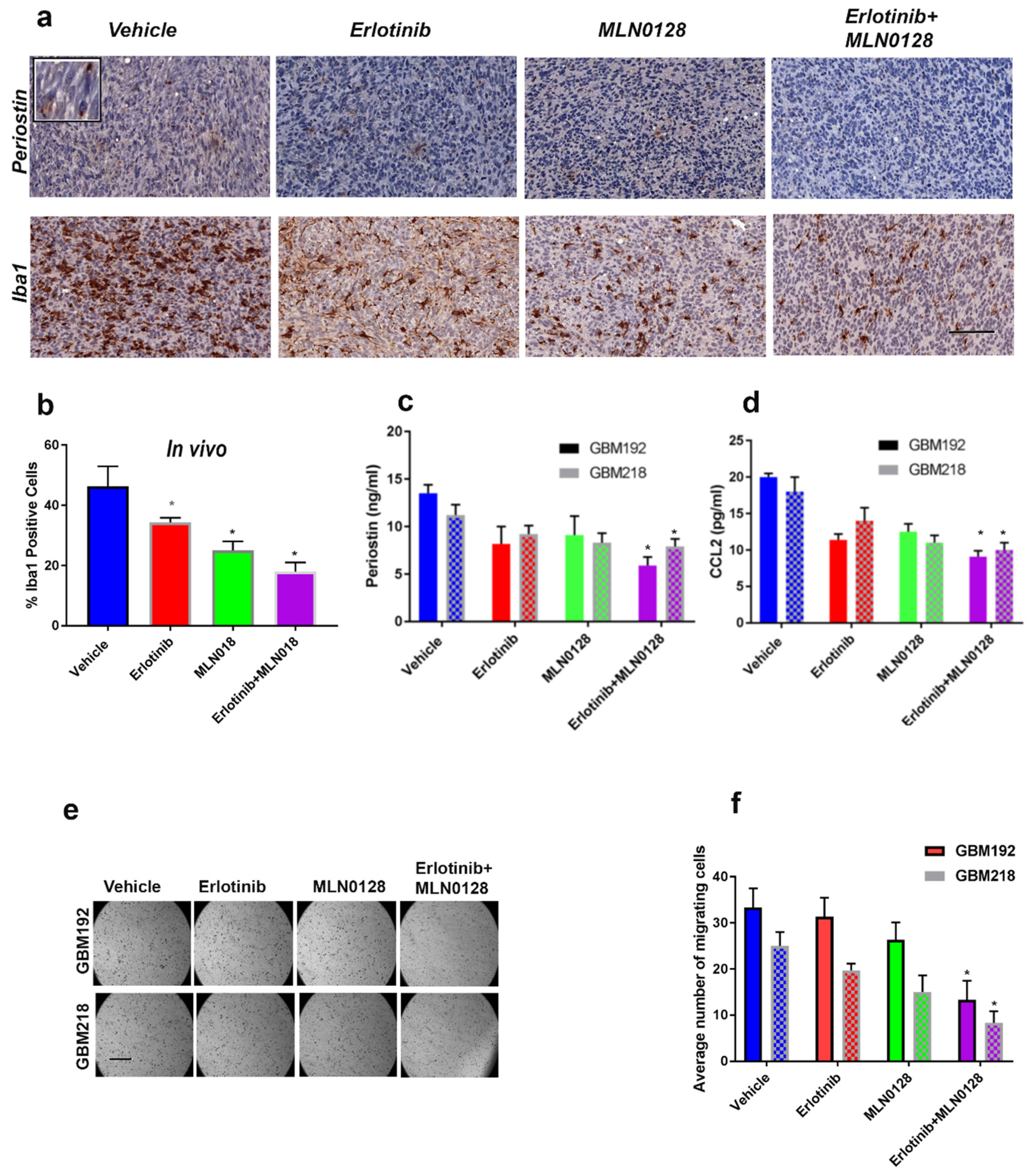
3.5. Combinatorial Therapy Using Erlotinib and MLN0128 Downregulates POSTN and CCL2 Levels and Inhibits the Iba1-Positive Macrophages in the GBM TME
4. Discussion
5. Future Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Singapore, 2017; Chapter 8. [Google Scholar]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (>/= 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef] [PubMed]
- Sadahiro, H.; Kang, K.D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro. Oncol. 2010, 12, 882–889. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Saleem, H.; Kulsoom Abdul, U.; Kucukosmanoglu, A.; Houweling, M.; Cornelissen, F.M.G.; Heiland, D.H.; Hegi, M.E.; Kouwenhoven, M.C.M.; Bailey, D.; Wurdinger, T.; et al. The TICking clock of EGFR therapy resistance in glioblastoma: Target Independence or target Compensation. Drug Resist. Updates 2019, 43, 29–37. [Google Scholar] [CrossRef]
- Boch, T.; Kohler, J.; Janning, M.; Loges, S. Targeting the EGF receptor family in non-small cell lung cancer-increased complexity and future perspectives. Cancer Biol. Med. 2022, 19, 1543–1564. [Google Scholar] [CrossRef]
- Chan, D.L.H.; Segelov, E.; Wong, R.S.; Smith, A.; Herbertson, R.A.; Li, B.T.; Tebbutt, N.; Price, T.; Pavlakis, N. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane. Database Syst. Rev. 2017, 6, CD007047. [Google Scholar] [CrossRef]
- Hu, C.; Leche, C.A., 2nd; Kiyatkin, A.; Yu, Z.; Stayrook, S.E.; Ferguson, K.M.; Lemmon, M.A. Glioblastoma mutations alter EGFR dimer structure to prevent ligand bias. Nature 2022, 602, 518–522. [Google Scholar] [CrossRef]
- Yung, W.K.; Vredenburgh, J.J.; Cloughesy, T.F.; Nghiemphu, P.; Klencke, B.; Gilbert, M.R.; Reardon, D.A.; Prados, M.D. Safety and efficacy of erlotinib in first-relapse glioblastoma: A phase II open-label study. Neuro Oncol. 2010, 12, 1061–1070. [Google Scholar] [CrossRef]
- Gallego, O.; Cuatrecasas, M.; Benavides, M.; Segura, P.P.; Berrocal, A.; Erill, N.; Colomer, A.; Quintana, M.J.; Balana, C.; Gil, M.; et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J. Neurooncol. 2014, 116, 413–419. [Google Scholar] [CrossRef]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; DeSilva, A.A.; Rabbitt, J.E.; Prados, M.D. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro Oncol. 2014, 16, 984–990. [Google Scholar] [CrossRef]
- Byron, S.A.; Tran, N.L.; Halperin, R.F.; Phillips, J.J.; Kuhn, J.G.; de Groot, J.F.; Colman, H.; Ligon, K.L.; Wen, P.Y.; Cloughesy, T.F.; et al. Prospective Feasibility Trial for Genomics-Informed Treatment in Recurrent and Progressive Glioblastoma. Clin. Cancer Res. 2018, 24, 295–305. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Lee, E.Q.; Nayak, L.; Wen, P.Y.; Reardon, D.A. Treatment options in newly diagnosed glioblastoma. Curr. Treat Options Neurol. 2013, 15, 281–288. [Google Scholar] [CrossRef]
- Chen, S.; Le, T.; Harley, B.A.C.; Imoukhuede, P.I. Characterizing Glioblastoma Heterogeneity via Single-Cell Receptor Quantification. Front. Genet. 2018, 6, 92. [Google Scholar] [CrossRef]
- Ketchen, S.E.; Gamboa-Esteves, F.O.; Lawler, S.E.; Nowicki, M.O.; Rohwedder, A.; Knipp, S.; Prior, S.; Short, S.C.; Ladbury, J.E.; Bruning-Richardson, A. Drug Resistance in Glioma Cells Induced by a Mesenchymal-Amoeboid Migratory Switch. Biomedicines 2021, 10, 9. [Google Scholar] [CrossRef]
- Ice, R.J.; Chen, M.; Sidorov, M.; Le Ho, T.; Woo, R.W.L.; Rodriguez-Brotons, A.; Luu, T.; Jian, D.; Kim, K.B.; Leong, S.P.; et al. Drug responses are conserved across patient-derived xenograft models of melanoma leading to identification of novel drug combination therapies. Br. J. Cancer 2020, 122, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Matlaf, L.; Khan, S.; Akhavan, A.; Singer, E.; Bezrookove, V.; Decker, S.; Ghanny, S.; Hadaczek, P.; Bengtsson, H.; et al. Cytomegalovirus Immediate-Early Proteins Promote Stemness Properties in Glioblastoma. Cancer Res. 2015, 75, 3065–3076. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Singer, E.; Dighe, P.; Sidorov, M.; Limbad, C.; Rodriquez-Brotons, A.; Rix, P.; Woo, R.W.L.; Dickinson, L.; Desprez, P.Y.; et al. Cannabidiol inhibits RAD51 and sensitizes glioblastoma to temozolomide in multiple orthotopic tumor models. Neurooncol. Adv. 2022, 4, vdac019. [Google Scholar] [CrossRef] [PubMed]
- Zusman, E.; Sidorov, M.; Ayala, A.; Chang, J.; Singer, E.; Chen, M.; Desprez, P.Y.; McAllister, S.; Salomonis, N.; Chetal, K.; et al. Tissues Harvested Using an Automated Surgical Approach Confirm Molecular Heterogeneity of Glioblastoma and Enhance Specimen’s Translational Research Value. Front. Oncol. 2019, 9, 1119. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Bezrookove, V.; Khan, I.A.; Nosrati, M.; Miller, J.R., 3rd; McAllister, S.; Dar, A.A.; Kashani-Sabet, M. BPTF promotes the progression of distinct subtypes of breast cancer and is a therapeutic target. Front. Oncol. 2022, 12, 1011173. [Google Scholar] [CrossRef]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Amp Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Eskilsson, E.; Rosland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018, 20, 743–752. [Google Scholar] [CrossRef]
- Zhang, N.; Fu, J.N.; Chou, T.C. Synergistic combination of microtubule targeting anticancer fludelone with cytoprotective panaxytriol derived from panax ginseng against MX-1 cells in vitro: Experimental design and data analysis using the combination index method. Am. J. Cancer Res. 2016, 6, 97–104. [Google Scholar]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- An, Z.; Knobbe-Thomsen, C.B.; Wan, X.; Fan, Q.W.; Reifenberger, G.; Weiss, W.A. EGFR Cooperates with EGFRvIII to Recruit Macrophages in Glioblastoma. Cancer Res. 2018, 78, 6785–6794. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Lim, M.; Puttick, S.; Houston, Z.H.; Thurecht, K.J.; Kalita-de Croft, P.; Mahler, S.; Rose, S.E.; Jeffree, R.L.; Mazzieri, R.; Dolcetti, R.; et al. Innovative Therapeutic Strategies for Effective Treatment of Brain Metastases. Int. J. Mol. Sci. 2019, 20, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, A.; Liffers, K.; Kathagen, A.; Riethdorf, S.; Zapf, S.; Merlo, A.; Kolbe, K.; Westphal, M.; Lamszus, K. Erlotinib resistance in EGFR-amplified glioblastoma cells is associated with upregulation of EGFRvIII and PI3Kp110delta. Neuro Oncol. 2013, 15, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Preusser, M. In search of a target: PD-1 and PD-L1 profiling across glioma types. Neuro Oncol. 2016, 18, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.J.; Chen, J.S.; Jain, S.; Morshed, R.A.; Haddad, A.F.; Gill, S.; Beniwal, A.S.; Aghi, M.K. Immunotherapy Resistance in Glioblastoma. Front. Genet. 2021, 12, 750675. [Google Scholar] [CrossRef]
- Shao, R. YKL-40 acts as an angiogenic factor to promote tumor angiogenesis. Front. Physiol. 2013, 4, 122. [Google Scholar] [CrossRef]
- Chen, A.; Jiang, Y.; Li, Z.; Wu, L.; Santiago, U.; Zou, H.; Cai, C.; Sharma, V.; Guan, Y.; McCarl, L.H.; et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J. Clin. Investig. 2021, 131, e147552. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, Z.; Sa, J.K.; Shin, S.; Wang, J.; Bordyuh, M.; Cho, H.J.; Elliott, O.; Chu, T.; Choi, S.W.; et al. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 2018, 50, 1399–1411. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Clone and Isotype | Dilution | Catalog Number | Supplier |
|---|---|---|---|---|
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | D13.14.4E; IgG | 1:1000 | 4370S | Cell Signaling Technology |
| p44/42 MAPK (Erk1/2) | 137F5; IgG | 1:1000 | 4695S | Cell Signaling Technology |
| Phospho-Stat3 (Tyr705) | D3A7; IgG | 1:500 | 9145S | Cell Signaling Technology |
| STAT3 | polyclonal | 1:1000 | 9132S | Cell Signaling Technology |
| Phospho-EGFR (Tyr1068) | polyclonal | 1:500 | 44-788G | Invitrogen, Waltham, MA, USA |
| EGF Receptor (T43) | polyclonal | 1:500 | 2963S | Cell Signaling Technology |
| Phospho-Akt (Ser473) | polyclonal | 1:500 | 9271S | Cell Signaling Technology |
| Akt | polyclonal | 1:1000 | 9272S | Cell Signaling Technology |
| EGFRvIII | DH8.3 | 1:500 | NBP2-50599 | Novus Biologicals, Centennial, CO, USA |
| GAPDH | 6C5, IgG1 | 1:5000 | MAB374 | Millipore-Sigma, Burlington, MA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorov, M.; Dighe, P.; Woo, R.W.L.; Rodriguez-Brotons, A.; Chen, M.; Ice, R.J.; Vaquero, E.; Jian, D.; Desprez, P.-Y.; Nosrati, M.; et al. Dual Targeting of EGFR and MTOR Pathways Inhibits Glioblastoma Growth by Modulating the Tumor Microenvironment. Cells 2023, 12, 547. https://doi.org/10.3390/cells12040547
Sidorov M, Dighe P, Woo RWL, Rodriguez-Brotons A, Chen M, Ice RJ, Vaquero E, Jian D, Desprez P-Y, Nosrati M, et al. Dual Targeting of EGFR and MTOR Pathways Inhibits Glioblastoma Growth by Modulating the Tumor Microenvironment. Cells. 2023; 12(4):547. https://doi.org/10.3390/cells12040547
Chicago/Turabian StyleSidorov, Maxim, Pratiksha Dighe, Rinette W. L. Woo, Aida Rodriguez-Brotons, Michelle Chen, Ryan J. Ice, Edith Vaquero, Damon Jian, Pierre-Yves Desprez, Mehdi Nosrati, and et al. 2023. "Dual Targeting of EGFR and MTOR Pathways Inhibits Glioblastoma Growth by Modulating the Tumor Microenvironment" Cells 12, no. 4: 547. https://doi.org/10.3390/cells12040547