
Establishment and Molecular Characterization of Two Patient-Derived Pancreatic Ductal Adenocarcinoma Cell Lines as Preclinical Models for Treatment Response
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
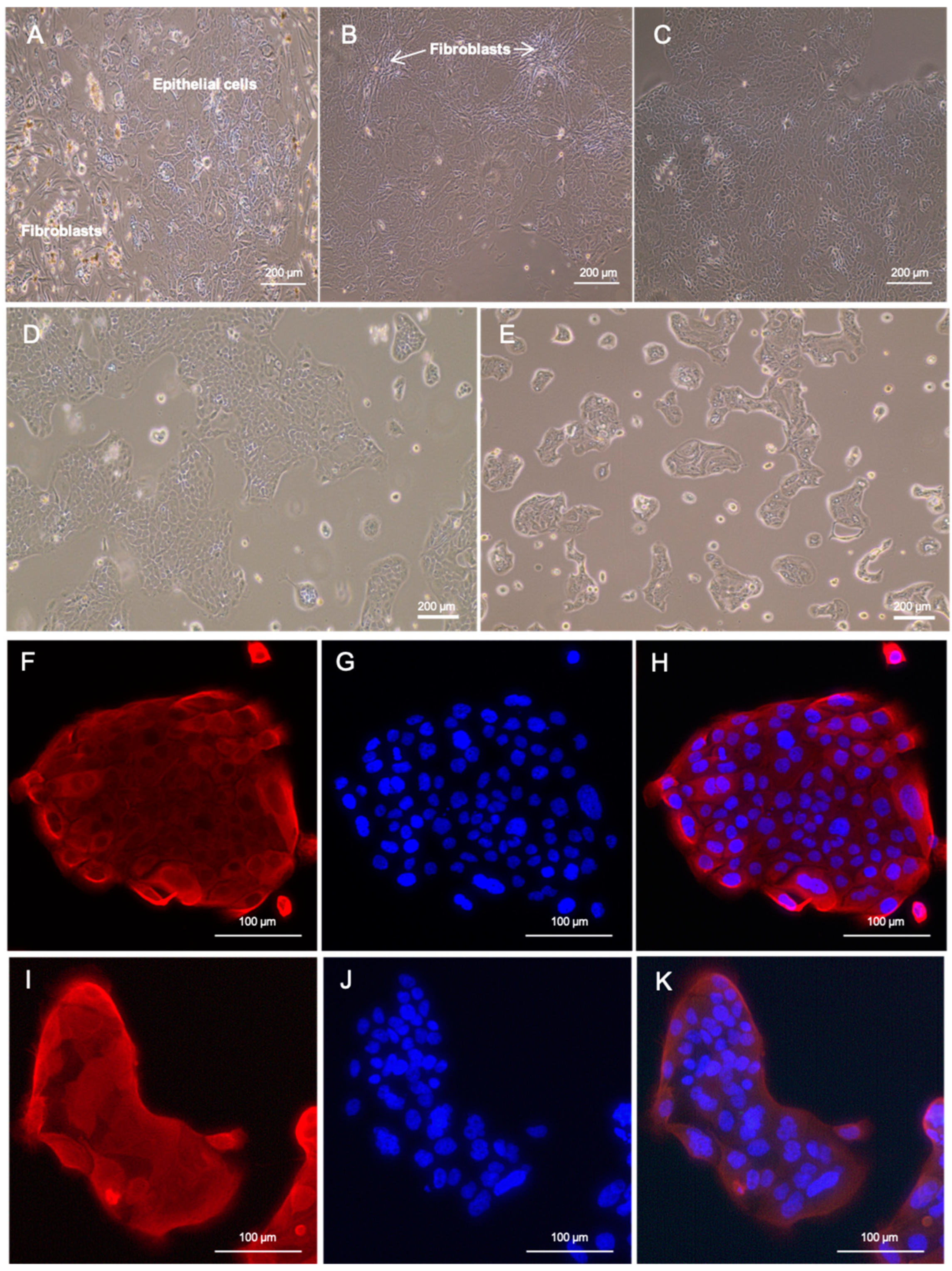
2.1. Tissue Collection, Cell Line Establishment, Fibroblast Depletion, and Generation of Single-Cell-Derived Clonal Cultures
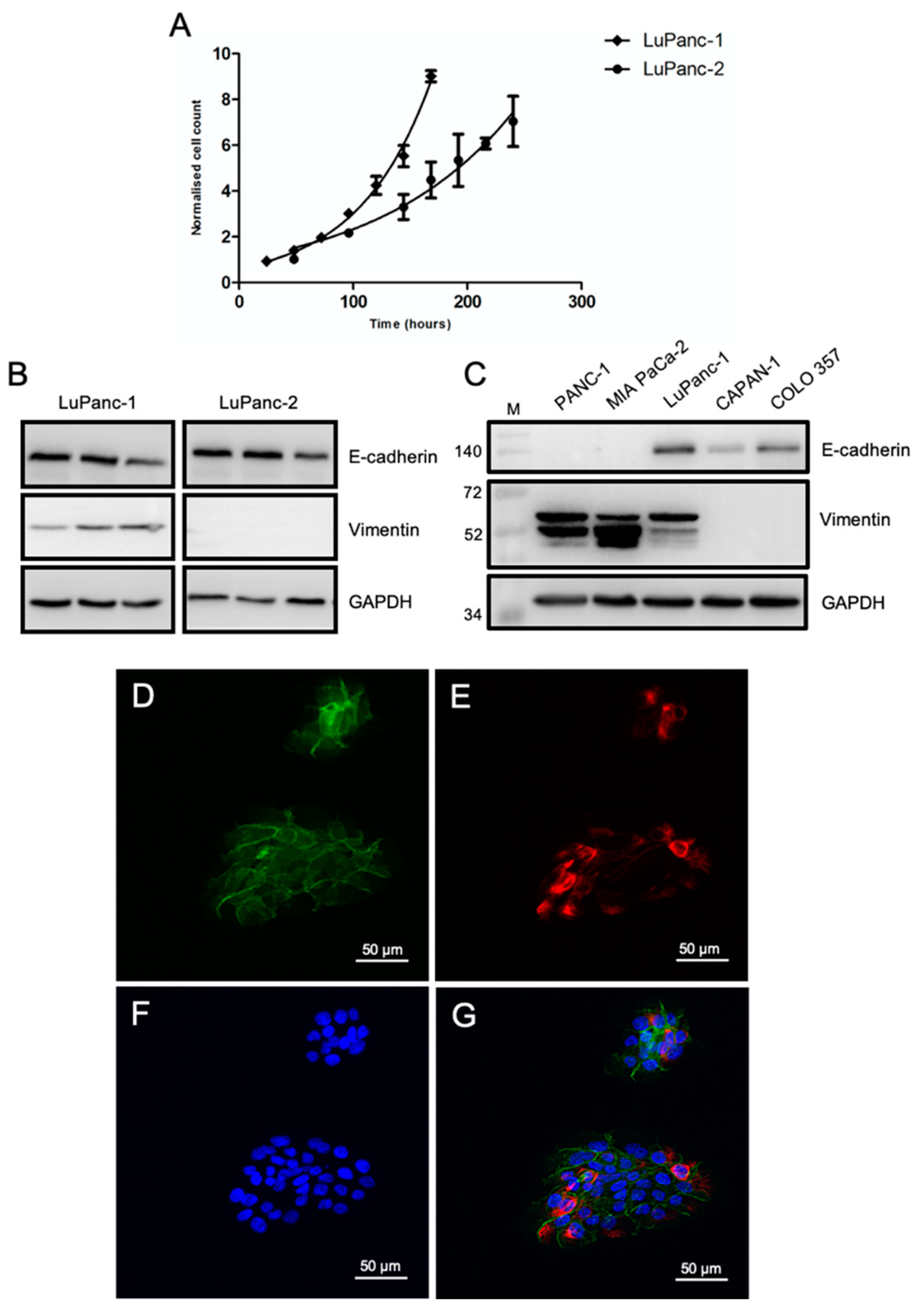
2.2. Growth Curves
2.3. Chemotherapy and Measurement of Cell Survival
2.4. Immunofluorescence
2.5. Immunoblotting
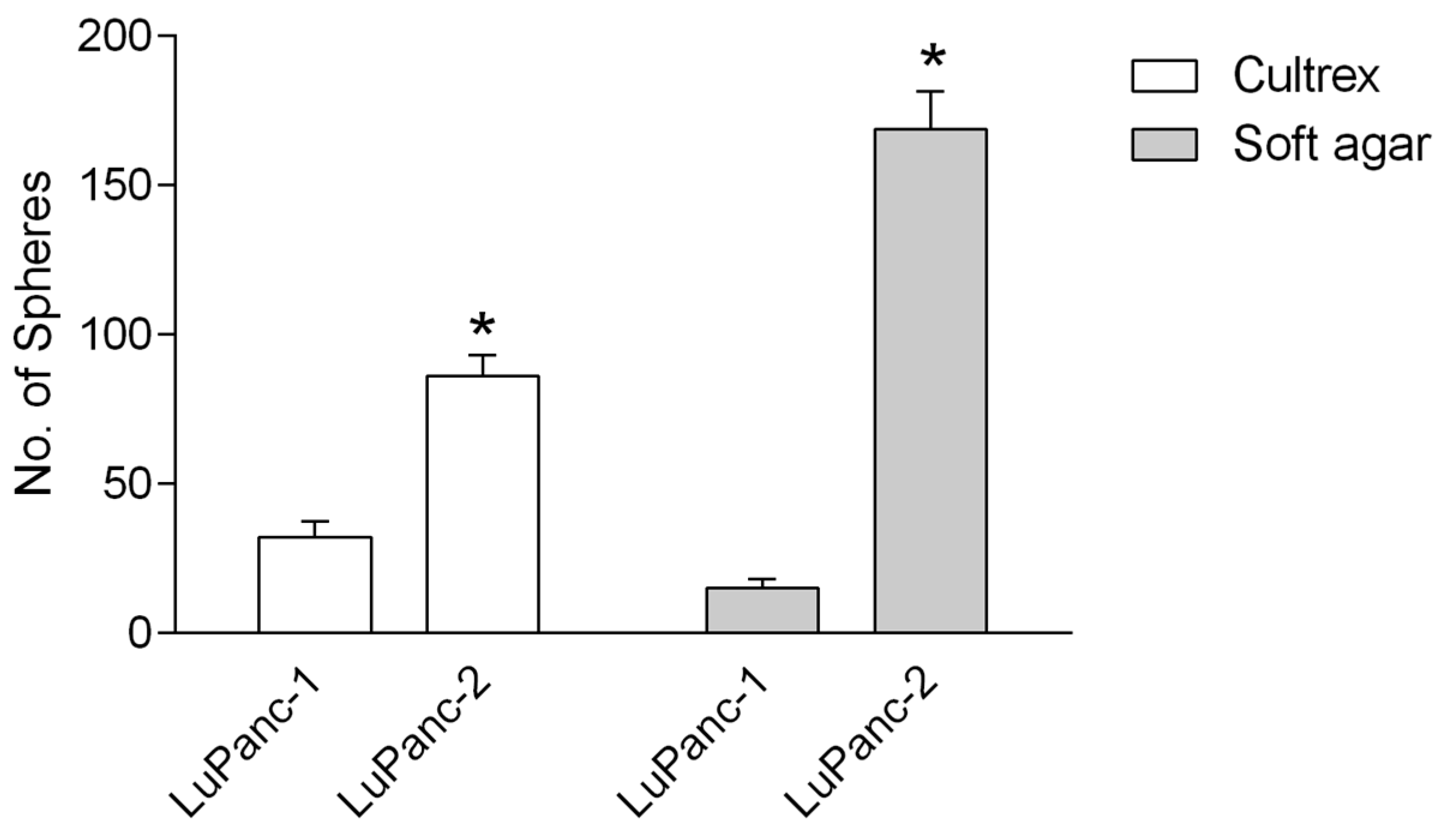
2.6. Soft Agar/Matrigel-Based Sphere-Forming Assays
2.7. Migration Assays
2.8. Whole Exome and Sanger Sequencing
2.9. Quantitative RT-PCR Analysis (qPCR)
3. Results
3.1. Patient History and the Establishment of Patient-Derived Cell Lines
3.2. Authentication of LuPanc-1 and LuPanc-2
3.3. Growth Kinetics and Epithelial-Mesenchymal Phenotyping
3.4. Genomic Landscape
3.5. Sphere-Forming Capacity in Semi-Solid Medium
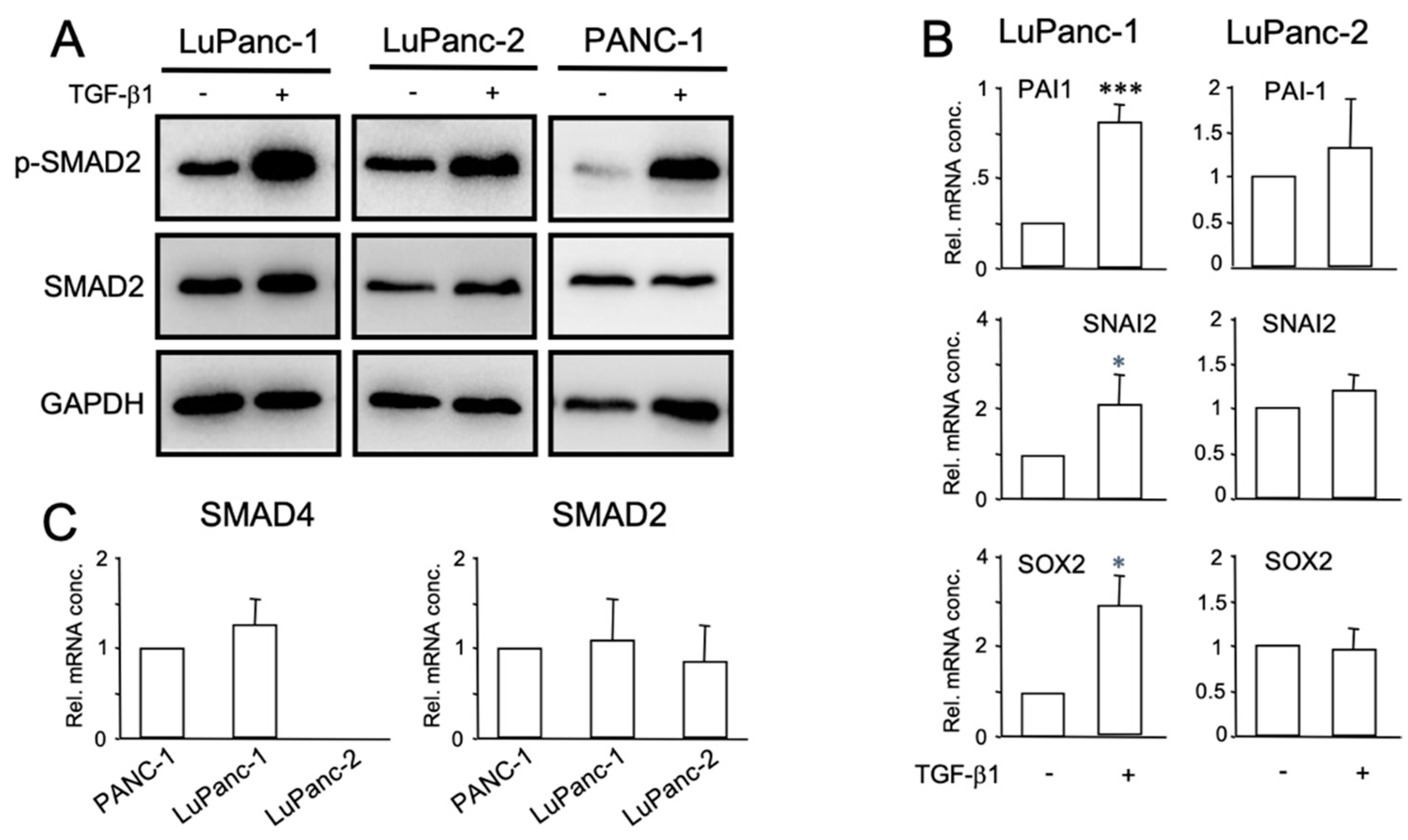
3.6. Characterization of TGF-β Signaling
3.7. Migratory Capacity
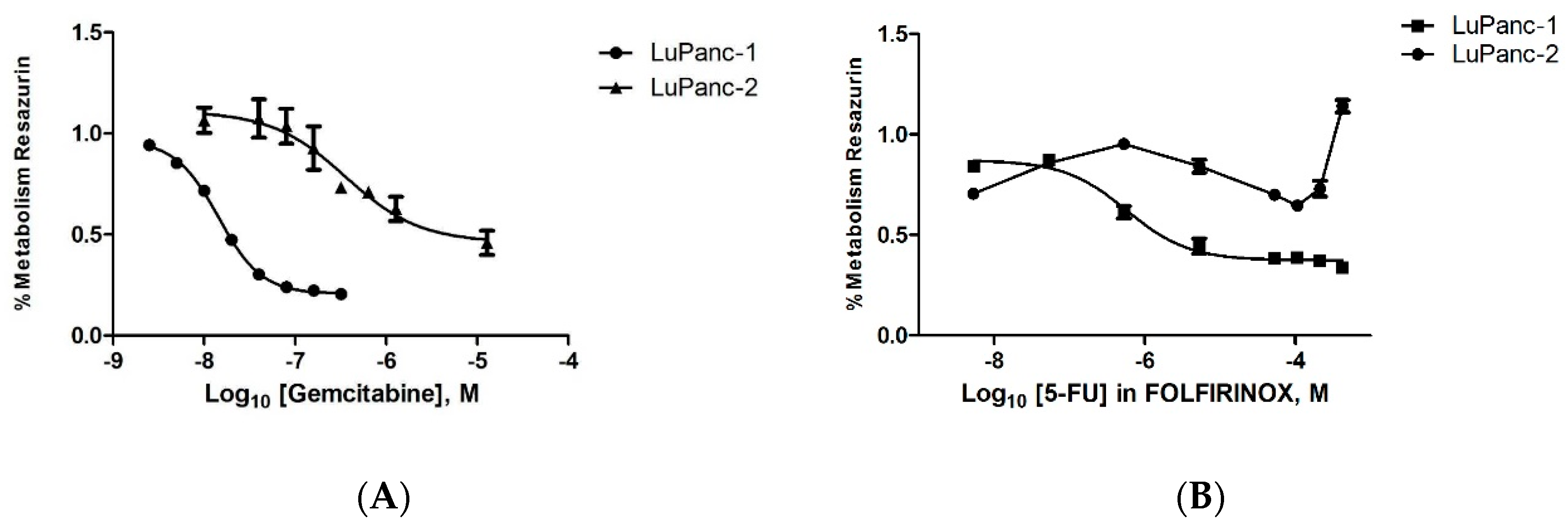
3.8. Treatment Response to Gemcitabine and FOLFIRINOX
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Han, Y.; Lee, H.; Kim, S.W.; Kwon, W.; Lee, K.H.; Oh, D.Y.; Chie, E.K.; Lee, J.M.; Heo, J.S.; et al. Oncological Benefits of Neoadjuvant Chemoradiation With Gemcitabine Versus Upfront Surgery in Patients With Borderline Resectable Pancreatic Cancer: A Prospective, Randomized, Open-label, Multicenter Phase 2/3 Trial. Ann. Surg. 2018, 268, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Janssen, Q.P.; Buettner, S.; Suker, M.; Beumer, B.R.; Addeo, P.; Bachellier, P.; Bahary, N.; Bekaii-Saab, T.; Bali, M.A.; Besselink, M.G.; et al. Neoadjuvant FOLFIRINOX in Patients With Borderline Resectable Pancreatic Cancer: A Systematic Review and Patient-Level Meta-Analysis. J. Natl. Cancer Inst. 2019, 111, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Janssen, Q.P.; O’Reilly, E.M.; van Eijck, C.H.J.; Groot Koerkamp, B. Neoadjuvant Treatment in Patients With Resectable and Borderline Resectable Pancreatic Cancer. Front. Oncol. 2020, 10, 41. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Heinemann, V.; Schmidberger, H. Duktales Adenokarzinom des Pankreas. Der. Onkol. 2019, 25, 642–644. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J.. Natl.. Cancer Inst.. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchene, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-betaRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers 2018, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.Y.; Liu, T.P.; Chou, C.J.; Chen, H.Y.; Lee, K.H.; Yang, P.M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Heinrich, K.; Böck, S.; Heinemann, V. Molekulare Charakterisierung des Pankreaskarzinoms. Der. Onkol. 2019, 25, 704–709. [Google Scholar] [CrossRef]
- Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1-A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. [Google Scholar] [CrossRef]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Fiedler, C.; Gadeken, T.; Kaufmann, R.; Lehnert, H.; Gieseler, F.; Rauch, B.H. The Role of PAR2 in TGF-beta1-Induced ERK Activation and Cell Motility. Int. J. Mol. Sci. 2017, 18, 2776. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic. Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Shyr, C.; Tarailo-Graovac, M.; Gottlieb, M.; Lee, J.J.; van Karnebeek, C.; Wasserman, W.W. FLAGS, frequently mutated genes in public exomes. BMC Med. Genom. 2014, 7, 64. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Spielmann, M.; Kakar, N.; Tayebi, N.; Leettola, C.; Nurnberg, G.; Sowada, N.; Lupianez, D.G.; Harabula, I.; Flottmann, R.; Horn, D.; et al. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome. Res. 2016, 26, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Dirks, W.G.; MacLeod, R.A.; Nakamura, Y.; Kohara, A.; Reid, Y.; Milch, H.; Drexler, H.G.; Mizusawa, H. Cell line cross-contamination initiative: An interactive reference database of STR profiles covering common cancer cell lines. Int. J. Cancer 2010, 126, 303–304. [Google Scholar] [CrossRef]
- Sinha, A.; Cherba, D.; Bartlam, H.; Lenkiewicz, E.; Evers, L.; Barrett, M.T.; Haab, B.B. Mesenchymal-like pancreatic cancer cells harbor specific genomic alterations more frequently than their epithelial-like counterparts. Mol. Oncol. 2014, 8, 1253–1265. [Google Scholar] [CrossRef]
- Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.U.; Ungefroren, H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers 2021, 13, 4663. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e.; Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203 e113. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Hwang, J.S.; Hong, Y.B.; Bae, I.; Seong, Y.S. Transforming growth factor beta receptor I inhibitor sensitizes drug-resistant pancreatic cancer cells to gemcitabine. Anticancer. Res. 2012, 32, 799–806. [Google Scholar]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Atienza, J.M.; Yu, N.; Kirstein, S.L.; Xi, B.; Wang, X.; Xu, X.; Abassi, Y.A. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev.. Technol. 2006, 4, 597–607. [Google Scholar] [CrossRef]
- Zuckerman, D.S.; Ryan, D.P. Adjuvant therapy for pancreatic cancer: A review. Cancer 2008, 112, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Neoptolemos, J.; Jager, D.; Buchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Ciliberto, D.; Staropoli, N.; Chiellino, S.; Botta, C.; Tassone, P.; Tagliaferri, P. Systematic review and meta-analysis on targeted therapy in advanced pancreatic cancer. Pancreatology 2016, 16, 249–258. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Finetti, P.; Birnbaum, D.; Mamessier, E.; Bertucci, F. Validation and comparison of the molecular classifications of pancreatic carcinomas. Mol. Cancer 2017, 16, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef]
- Richter, M.; Piwocka, O.; Musielak, M.; Piotrowski, I.; Suchorska, W.M.; Trzeciak, T. From Donor to the Lab: AFascinating Journey of Primary Cell Lines. Front. Cell Dev. Biol. 2021, 9, 711381. [Google Scholar] [CrossRef]
- Braun, R.; Anthuber, L.; Hirsch, D.; Wangsa, D.; Lack, J.; McNeil, N.E.; Heselmeyer-Haddad, K.; Torres, I.; Wangsa, D.; Brown, M.A.; et al. Single-Cell-Derived Primary Rectal Carcinoma Cell Lines Reflect Intratumor Heterogeneity Associated with Treatment Response. Clin. Cancer Res. 2020, 26, 3468–3480. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Saha, P.; Samanta, A.; Bishayee, A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules 2020, 10, 1561. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Groth, S.; Sebens Müerköster, S.; Sipos, B.; Schäfer, H.; Kalthoff, H.; Fändrich, F.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef] [PubMed]
- Schmidtlein, P.M.; Volz, C.; Hackel, A.; Thürling, I.; Castven, D.; Braun, R.; Wellner, U.F.; Konukiewitz, B.; Riemekasten, G.; Lehnert, H.; et al. Activation of a Ductal-to-Endocrine Transdifferentiation Transcriptional Program in the Pancreatic Cancer Cell Line PANC-1 Is Controlled by RAC1 and RAC1b through Antagonistic Regulation of Stemness Factors. Cancers 2021, 13, 5541. [Google Scholar] [CrossRef]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Liu, S.; Kleeff, J.; Friess, H.; Büchler, M.W. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology 2002, 62, 354–362. [Google Scholar] [CrossRef]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 2004, 10, 2307–2318. [Google Scholar] [CrossRef]
- Giovannetti, E.; Mey, V.; Danesi, R.; Mosca, I.; Del Tacca, M. Synergistic cytotoxicity and pharmacogenetics of gemcitabine and pemetrexed combination in pancreatic cancer cell lines. Clin. Cancer Res. 2004, 10, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Mori, R.; Ishikawa, T.; Ichikawa, Y.; Taniguchi, K.; Matsuyama, R.; Ueda, M.; Fujii, Y.; Endo, I.; Togo, S.; Danenberg, P.V.; et al. Human equilibrative nucleoside transporter 1 is associated with the chemosensitivity of gemcitabine in human pancreatic adenocarcinoma and biliary tract carcinoma cells. Oncol. Rep. 2007, 17, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Begg, S.K.S.; Birnbaum, D.J.; Clark, J.W.; Mino-Kenudson, M.; Wellner, U.F.; Schilling, O.; Lillemoe, K.D.; Warshaw, A.L.; Castillo, C.F.; Liss, A.S. FOLFIRINOX Versus Gemcitabine-based Therapy for Pancreatic Ductal Adenocarcinoma: Lessons from Patient-derived Cell Lines. Anticancer. Res. 2020, 40, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.; Ricigliano, M.; McCarthy, B.; Chou, J.F.; Capanu, M.; Cooper, B.; Bartlett, A.; Covington, C.; Lowery, M.A.; O’Reilly, E.M. Circulating Tumor and Invasive Cell Gene Expression Profile Predicts Treatment Response and Survival in Pancreatic Adenocarcinoma. Cancers 2018, 10, 467. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, S.C.; Hong, S.M.; Kim, Y.H.; Song, K.B.; Park, K.M.; Lee, Y.J. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas 2013, 42, 216–222. [Google Scholar] [CrossRef]
- Hsieh, H.H.S.; Agarwal, S.; Cholok, D.J.; Loder, S.J.; Kaneko, K.; Huber, A.; Chung, M.T.; Ranganathan, K.; Habbouche, J.; Li, J.; et al. Coordinating Tissue Regeneration Through Transforming Growth Factor-beta Activated Kinase 1 Inactivation and Reactivation. Stem Cells 2019, 37, 766–778. [Google Scholar] [CrossRef]
- Cui, Y.; Brosnan, J.A.; Blackford, A.L.; Sur, S.; Hruban, R.H.; Kinzler, K.W.; Vogelstein, B.; Maitra, A.; Diaz, L.A., Jr.; Iacobuzio-Donahue, C.A.; et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin. Cancer Res. 2012, 18, 6519–6530. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cancer Cell Line Encyclopedia, C.; Genomics of Drug Sensitivity in Cancer, C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Seo, Y.D.; Chang, J.H.; Coveler, A.; Nigjeh, E.N.; Pan, S.; Jalikis, F.; Yeung, R.S.; Crispe, I.N.; Pillarisetty, V.G. Long-lived pancreatic ductal adenocarcinoma slice cultures enable precise study of the immune microenvironment. Oncoimmunology 2017, 6, e1333210. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | LuPanc-1 | LuPanc-2 |
|---|---|---|
| D3S1358 | 16/17 | 15,17 |
| D1S1656 | 11/14 | 12,15 |
| D6S1043 | 12/17 | 19 |
| D13S317 | 11/12 | 11/12 |
| Penta E | 11 | 7/12 |
| D16S539 | 11/13 | 12,13 |
| D18S51 | 16 | 13 |
| D2S1338 | 22/23 | 17/23 |
| SCF1PO | 10/12 | 10/11 |
| Penta D | 13/15 | 10 |
| TH01 | 7/9 | 6/9 |
| vWA | 17/20 | 14/19 |
| D21S11 | 30 | 27/30 |
| D7S820 | 11 | 8/10 |
| D5S818 | 11/13 | 13 |
| TPOX | 12 | 8 |
| D8S1179 | 12/13 | 13/15 |
| D12S391 | 21/23 | 17/19 |
| D19S433 | 14/15 | 15 |
| FGA | 22/24 | 20/21.2 |
| AM | X/Y | X |
| Frame Shift Deletion | Frame Shift Insertion | In-Frame Deletion | Missense Mutation | Nonsense Mutation | Splice Site | Total | |
|---|---|---|---|---|---|---|---|
| LuPanc-1 | 0 | 1 | 2 | 124 | 5 | 3 | 135 |
| LuPanc-2 | 1 | 0 | 0 | 70 | 4 | 1 | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, R.; Lapshyna, O.; Watzelt, J.; Drenckhan, M.; Künstner, A.; Färber, B.; Hael, A.A.M.; Bolm, L.; Honselmann, K.C.; Konukiewitz, B.; et al. Establishment and Molecular Characterization of Two Patient-Derived Pancreatic Ductal Adenocarcinoma Cell Lines as Preclinical Models for Treatment Response. Cells 2023, 12, 587. https://doi.org/10.3390/cells12040587
Braun R, Lapshyna O, Watzelt J, Drenckhan M, Künstner A, Färber B, Hael AAM, Bolm L, Honselmann KC, Konukiewitz B, et al. Establishment and Molecular Characterization of Two Patient-Derived Pancreatic Ductal Adenocarcinoma Cell Lines as Preclinical Models for Treatment Response. Cells. 2023; 12(4):587. https://doi.org/10.3390/cells12040587
Chicago/Turabian StyleBraun, Rüdiger, Olha Lapshyna, Jessica Watzelt, Maren Drenckhan, Axel Künstner, Benedikt Färber, Ahmed Ahmed Mohammed Hael, Louisa Bolm, Kim Christin Honselmann, Björn Konukiewitz, and et al. 2023. "Establishment and Molecular Characterization of Two Patient-Derived Pancreatic Ductal Adenocarcinoma Cell Lines as Preclinical Models for Treatment Response" Cells 12, no. 4: 587. https://doi.org/10.3390/cells12040587