
The Role of CD38 in the Pathogenesis of Cardiorenal Metabolic Disease and Aging, an Approach from Basic Research
Abstract
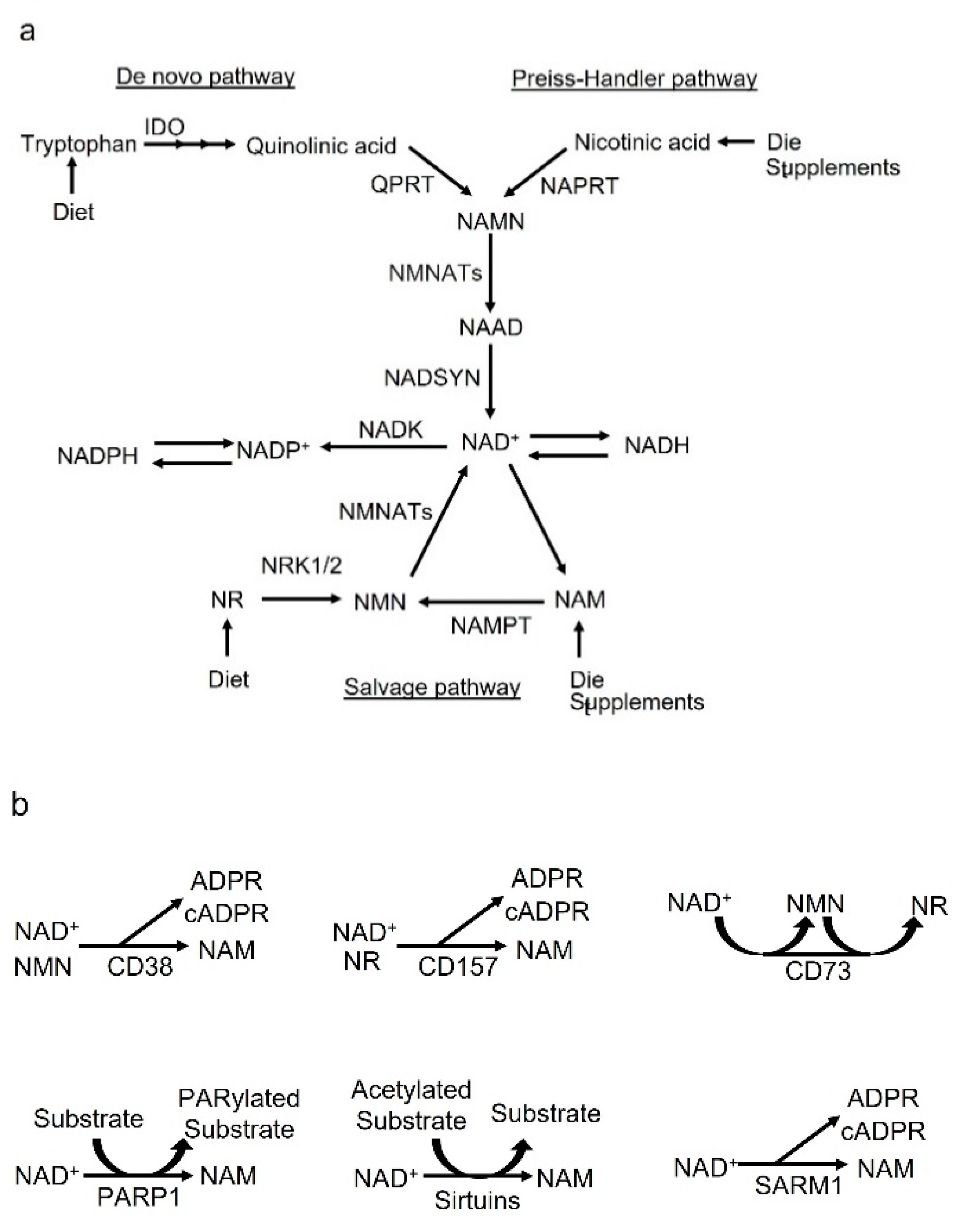
:1. Introduction
2. NAD+ Synthesis and Metabolic Cycle
2.1. De Novo Pathway and Preiss–Handler Pathway
2.2. Salvage Pathway
3. Biology of CD38
4. Role of CD38 on Aging, Metabolic Dysfunction, High-Fat-Diet-Induced Obesity and Insulin Secretion
5. Role of CD38 on Cardiovascular Disease and Kidney Disease
5.1. CD38 Gene Ablation or Inhibition Protects the Heart
5.2. CD38 Gene Ablation or Inhibition of Its Activation Protects Vascular Tissue and Cells
5.3. CD38 Gene Ablation or Inhibition of Its Activation Promotes Coronary Atherosclerosis
5.4. CD38 Plays a Crucial Role in Vasoconstriction
5.5. Role of CD38 on Kidney Disease
6. Conclusions and Prospectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Denic, A.; Glassock, R.J.; Rule, A.D. Structural and Functional Changes With the Aging Kidney. Adv. Chronic Kidney Dis. 2016, 23, 19–28. [Google Scholar] [CrossRef]
- Dominguez, L.J.; Barbagallo, M. The biology of the metabolic syndrome and aging. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Tas, E.; Yakar, S.; Muzumdar, R. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol. Cell. Endocrinol. 2017, 455, 115–130. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2016, 594, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, J.; Mu, X.; McGowan, S.J.; Angelini, L.; O’Kelly, R.D.; Yousefzadeh, M.J.; Sakamoto, A.; Aversa, Z.; LeBrasseur, N.K.; et al. Novel small molecule inhibition of IKK/NF-κB activation reduces markers of senescence and improves healthspan in mouse models of aging. Aging Cell 2021, 20, e13486. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-kappaB activation: From bench to bedside. Exp. Biol. Med. 2008, 233, 21–31. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell Mol. Life. Sci. 2012, 69, 2999–3013. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 2012, 7, e42357. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes. Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends. Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. 2013, 124, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Mauro, S.; Gévry, N.; Lis, J.T.; Kraus, W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef]
- Youn, H.S.; Kim, T.G.; Kim, M.K.; Kang, G.B.; Kang, J.Y.; Lee, J.G.; An, J.Y.; Park, K.R.; Lee, Y.; Im, Y.J.; et al. Structural Insights into the Quaternary Catalytic Mechanism of Hexameric Human Quinolinate Phosphoribosyltransferase, a Key Enzyme in de novo NAD Biosynthesis. Sci. Rep. 2016, 6, 19681. [Google Scholar] [CrossRef] [PubMed]
- Marletta, A.S.; Massarotti, A.; Orsomando, G.; Magni, G.; Rizzi, M.; Garavaglia, S. Crystal structure of human nicotinic acid phosphoribosyltransferase. FEBS OpenBio 2015, 5, 419–428. [Google Scholar] [CrossRef]
- Brazill, J.M.; Li, C.; Zhu, Y.; Zhai, R.G. NMNAT: It’s an NAD(+) synthase… It’s a chaperone… It’s a neuroprotector. Curr. Opin. Genet. Dev. 2017, 44, 156–162. [Google Scholar] [CrossRef]
- Rizzi, M.; Bolognesi, M.; Coda, A. A novel deamido-NAD+-binding site revealed by the trapped NAD-adenylate intermediate in the NAD+ synthetase structure. Structure 1998, 6, 1129–1140. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e1065. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.; Wolberger, C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, C.; Mazzola, F.; Raffaelli, N. The key role of the NAD biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase in regulating cell functions. IUBMB Life. 2022, 74, 562–572. [Google Scholar] [CrossRef]
- Buonvicino, D.; Mazzola, F.; Zamporlini, F.; Resta, F.; Ranieri, G.; Camaioni, E.; Muzzi, M.; Zecchi, R.; Pieraccini, G.; Dölle, C.; et al. Identification of the Nicotinamide Salvage Pathway as a New Toxification Route for Antimetabolites. Cell Chem. Biol. 2018, 25, 471–482.e477. [Google Scholar] [CrossRef] [PubMed]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 2018, 360, eaan5780. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Palikhe, S.; Izumi, H.; Yoshida, T.; Hikosaka, K.; Hayat, F.; Karim, M.; Iqbal, T.; Nitta, Y.; Sato, A.; et al. BST1 regulates nicotinamide riboside metabolism via its glycohydrolase and base-exchange activities. Nat. Commun. 2021, 12, 6767. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Katayama, S.; Ariyoshi, M.; Ishihara, K.; Hirano, T.; Jingami, H.; Morikawa, K. Crystallographic studies on human BST-1/CD157 with ADP-ribosyl cyclase and NAD glycohydrolase activities. J. Mol. Biol. 2002, 316, 711–723. [Google Scholar] [CrossRef]
- Jablonska, P.; Kutryb-Zajac, B.; Mierzejewska, P.; Jasztal, A.; Bocian, B.; Lango, R.; Rogowski, J.; Chlopicki, S.; Smolenski, R.T.; Slominska, E.M. The new insight into extracellular NAD(+) degradation-the contribution of CD38 and CD73 in calcific aortic valve disease. J. Cell Mol. Med. 2021, 25, 5884–5898. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, P.; Mierzejewska, P.; Tomczyk, M.; Koszalka, P.; Franczak, M.; Kawecka, A.; Kutryb-Zajac, B.; Braczko, A.; Smolenski, R.T.; Slominska, E.M. Differences in Extracellular NAD(+) and NMN Metabolism on the Surface of Vascular Endothelial Cells. Biology 2022, 11, 675. [Google Scholar] [CrossRef]
- Olsson, A.; Olofsson, T.; Pero, R.W. Specific binding and uptake of extracellular nicotinamide in human leukemic K-562 cells. Biochem. Pharmacol. 1993, 45, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Okuda, H.; Nishida, K.; Fujimoto, S.; Nagasawa, K. Protective effect of nicotinamide against poly(ADP-ribose) polymerase-1-mediated astrocyte death depends on its transporter-mediated uptake. Life Sci. 2010, 86, 676–682. [Google Scholar] [CrossRef]
- Nikiforov, A.; Dölle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011, 286, 21767–21778. [Google Scholar] [CrossRef] [PubMed]
- Sociali, G.; Raffaghello, L.; Magnone, M.; Zamporlini, F.; Emionite, L.; Sturla, L.; Bianchi, G.; Vigliarolo, T.; Nahimana, A.; Nencioni, A.; et al. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 2016, 7, 2968–2984. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Summers, D.W.; Gibson, D.A.; DiAntonio, A.; Milbrandt, J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc. Natl. Acad. Sci. USA 2016, 113, E6271–E6280. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e1335. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Graeff, R.; Liu, Q.; Kriksunov, I.A.; Kotaka, M.; Oppenheimer, N.; Hao, Q.; Lee, H.C. Mechanism of cyclizing NAD to cyclic ADP-ribose by ADP-ribosyl cyclase and CD38. J. Biol. Chem. 2009, 284, 27629–27636. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef]
- Gul, R.; Park, D.R.; Shawl, A.I.; Im, S.Y.; Nam, T.S.; Lee, S.H.; Ko, J.K.; Jang, K.Y.; Kim, D.; Kim, U.H. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Cyclic ADP-Ribose (cADPR) Mediate Ca2+ Signaling in Cardiac Hypertrophy Induced by β-Adrenergic Stimulation. PLoS ONE 2016, 11, e0149125. [Google Scholar] [CrossRef]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca(2+)-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in β-adrenoceptor signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef] [PubMed]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.; Kato, I.; Takasawa, S.; Okamoto, H. CD38 is the major enzyme responsible for synthesis of nicotinic acid-adenine dinucleotide phosphate in mammalian tissues. Biochem. J. 2002, 362, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Hohenegger, M.; Suko, J.; Gscheidlinger, R.; Drobny, H.; Zidar, A. Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem. J. 2002, 367, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Langhorst, M.F.; Schwarzmann, N.; Guse, A.H. Ca2+ release via ryanodine receptors and Ca2+ entry: Major mechanisms in NAADP-mediated Ca2+ signaling in T-lymphocytes. Cell Signal. 2004, 16, 1283–1289. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Zhu, Y.; Rossi, D.; Sorrentino, V.; Ma, J.; Zhu, M.X.; Evans, A.M. Cyclic adenosine diphosphate ribose activates ryanodine receptors, whereas NAADP activates two-pore domain channels. J. Biol. Chem. 2011, 286, 9136–9140. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Funnell, T.M.; Sitsapesan, M.; Venturi, E.; Rietdorf, K.; Ruas, M.; Ganesan, A.; Gosain, R.; Churchill, G.C.; Zhu, M.X.; et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J. Biol. Chem. 2010, 285, 35039–35046. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Y.J.; Li, W.H.; Hou, Y.N.; Li, T.; Zhao, Z.Y.; Fang, C.; Li, S.L.; Lee, H.C. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc. Natl. Acad. Sci. USA 2017, 114, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- Shrimp, J.H.; Hu, J.; Dong, M.; Wang, B.S.; MacDonald, R.; Jiang, H.; Hao, Q.; Yen, A.; Lin, H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 2014, 136, 5656–5663. [Google Scholar] [CrossRef]
- Sun, L.; Adebanjo, O.A.; Koval, A.; Anandatheerthavarada, H.K.; Iqbal, J.; Wu, X.Y.; Moonga, B.S.; Wu, X.B.; Biswas, G.; Bevis, P.J.; et al. A novel mechanism for coupling cellular intermediary metabolism to cytosolic Ca2+ signaling via CD38/ADP-ribosyl cyclase, a putative intracellular NAD+ sensor. FASEB J. 2002, 16, 302–314. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar] [CrossRef]
- Astigiano, C.; Benzi, A.; Laugieri, M.E.; Piacente, F.; Sturla, L.; Guida, L.; Bruzzone, S.; De Flora, A. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells 2022, 11, 2637. [Google Scholar] [CrossRef] [PubMed]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-de-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef]
- Musso, T.; Deaglio, S.; Franco, L.; Calosso, L.; Badolato, R.; Garbarino, G.; Dianzani, U.; Malavasi, F. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J. Leukoc. Biol. 2001, 69, 605–612. [Google Scholar] [CrossRef]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-κB signaling suppression. Cell Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef]
- Matalonga, J.; Glaria, E.; Bresque, M.; Escande, C.; Carbó, J.M.; Kiefer, K.; Vicente, R.; León, T.E.; Beceiro, S.; Pascual-García, M.; et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017, 18, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.N.; Tirumurugaan, K.G.; Deshpande, D.A.; Amrani, Y.; Panettieri, R.A.; Walseth, T.F.; Kannan, M.S. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: Role of NF-kappaB and sensitivity to glucocorticoids. Faseb. j. 2006, 20, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Tirumurugaan, K.G.; Jude, J.A.; Kang, B.N.; Panettieri, R.A.; Walseth, T.F.; Kannan, M.S. TNF-alpha induced CD38 expression in human airway smooth muscle cells: Role of MAP kinases and transcription factors NF-kappaB and AP-1. Am J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1385–L1395. [Google Scholar] [CrossRef]
- Guedes, A.G.; Deshpande, D.A.; Dileepan, M.; Walseth, T.F.; Panettieri, R.A., Jr.; Subramanian, S.; Kannan, M.S. CD38 and airway hyper-responsiveness: Studies on human airway smooth muscle cells and mouse models. Can. J. Physiol. Pharmacol. 2015, 93, 145–153. [Google Scholar] [CrossRef]
- Barata, H.; Thompson, M.; Zielinska, W.; Han, Y.S.; Mantilla, C.B.; Prakash, Y.S.; Feitoza, S.; Sieck, G.; Chini, E.N. The role of cyclic-ADP-ribose-signaling pathway in oxytocin-induced Ca2+ transients in human myometrium cells. Endocrinology 2004, 145, 881–889. [Google Scholar] [CrossRef]
- Lee, C.U.; Song, E.K.; Yoo, C.H.; Kwak, Y.K.; Han, M.K. Lipopolysaccharide induces CD38 expression and solubilization in J774 macrophage cells. Mol. Cells 2012, 34, 573–576. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, X.X.; Ritter, J.K.; Abais, J.M.; Zhang, Y.; Li, P.L. Contribution of NADPH oxidase to membrane CD38 internalization and activation in coronary arterial myocytes. PLoS ONE 2013, 8, e71212. [Google Scholar] [CrossRef] [PubMed]
- Tohgo, A.; Takasawa, S.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Furuya, Y.; Yonekura, H.; Okamoto, H. Essential cysteine residues for cyclic ADP-ribose synthesis and hydrolysis by CD38. J. Biol. Chem. 1994, 269, 28555–28557. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Kim, S.J.; Park, Y.R.; Shin, Y.M.; Park, H.J.; Park, K.J.; Park, K.H.; Kim, H.K.; Jang, S.I.; An, N.H.; et al. Antidiabetic effect of a prodrug of cysteine, L-2-oxothiazolidine-4-carboxylic acid, through CD38 dimerization and internalization. J. Biol. Chem. 2002, 277, 5315–5321. [Google Scholar] [CrossRef]
- Park, D.R.; Nam, T.S.; Kim, Y.W.; Bae, Y.S.; Kim, U.H. Oxidative activation of type III CD38 by NADPH oxidase-derived hydrogen peroxide in Ca(2+) signaling. FASEB J. 2019, 33, 3404–3419. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, H.; Wang, Q.; Deng, H. Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. J. Proteome Res. 2014, 13, 786–795. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Larrick, J.W.; Larrick, J.W.; Mendelsohn, A.R. Contribution of Ferroptosis to Aging and Frailty. Rejuvenation Res. 2020, 23, 434–438. [Google Scholar] [CrossRef]
- Ma, Y.; Yi, M.; Wang, W.; Liu, X.; Wang, Q.; Liu, C.; Chen, Y.; Deng, H. Oxidative degradation of dihydrofolate reductase increases CD38-mediated ferroptosis susceptibility. Cell Death. Dis. 2022, 13, 944. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef] [Green Version]
- Peclat, T.R.; Thompson, K.L.; Warner, G.M.; Chini, C.C.S.; Tarragó, M.G.; Mazdeh, D.Z.; Zhang, C.; Zavala-Solorio, J.; Kolumam, G.; Liang Wong, Y.; et al. CD38 inhibitor 78c increases mice lifespan and healthspan in a model of chronological aging. Aging Cell 2022, 21, e13589. [Google Scholar] [CrossRef]
- Polzonetti, V.; Carpi, F.M.; Micozzi, D.; Pucciarelli, S.; Vincenzetti, S.; Napolioni, V. Population variability in CD38 activity: Correlation with age and significant effect of TNF-α -308G>A and CD38 184C>G SNPs. Mol. Genet. Metab. 2012, 105, 502–507. [Google Scholar] [CrossRef]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Chiang, S.H.; Harrington, W.W.; Luo, G.; Milliken, N.O.; Ulrich, J.C.; Chen, J.; Rajpal, D.K.; Qian, Y.; Carpenter, T.; Murray, R.; et al. Genetic Ablation of CD38 Protects against Western Diet-Induced Exercise Intolerance and Metabolic Inflexibility. PLoS ONE 2015, 10, e0134927. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Miao, L.J.; Wang, X.N.; Huang, C.C.; Qian, Y.S.; Huang, X.; Wang, X.L.; Jin, W.Z.; Ji, G.J.; Fu, M.; et al. CD38 deficiency suppresses adipogenesis and lipogenesis in adipose tissues through activating Sirt1/PPARγ signaling pathway. J. Cell Mol. Med. 2018, 22, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013, 62, 1084–1093. [Google Scholar] [CrossRef]
- Xie, L.; Wen, K.; Li, Q.; Huang, C.C.; Zhao, J.L.; Zhao, Q.H.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Gan, L.; et al. CD38 Deficiency Protects Mice from High Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Activating NAD(+)/Sirtuins Signaling Pathways-Mediated Inhibition of Lipid Accumulation and Oxidative Stress in Hepatocytes. Int. J. Biol. Sci. 2021, 17, 4305–4315. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Kato, I.; Yamamoto, Y.; Fujimura, M.; Noguchi, N.; Takasawa, S.; Okamoto, H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 1999, 274, 1869–1872. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Ford, E.L.; Bernal-Mizrachi, E.; Kusser, K.L.; Luciani, D.S.; Han, Z.; Tran, H.; Randall, T.D.; Lund, F.E.; Polonsky, K.S. Suppressed insulin signaling and increased apoptosis in CD38-null islets. Diabetes 2006, 55, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Y.G.; Reifsnyder, P.C.; Schott, W.H.; Lee, C.H.; Osborne, M.; Scheuplein, F.; Haag, F.; Koch-Nolte, F.; Serreze, D.V.; et al. Targeted disruption of CD38 accelerates autoimmune diabetes in NOD/Lt mice by enhancing autoimmunity in an ADP-ribosyltransferase 2-dependent fashion. J. Immunol. 2006, 176, 4590–4599. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Euler, G.; Schulz, R. Vascular and Cardiac Oxidative Stress and Inflammation as Targets for Cardioprotection. Curr. Pharm. Des. 2021, 27, 2112–2130. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Jiang, W.; Gan, L.; Wang, L.; Sun, C.; Ni, P.; Liu, Y.; Wu, S.; Gu, L.; Zheng, W.; et al. Mouse embryonic fibroblasts from CD38 knockout mice are resistant to oxidative stresses through inhibition of reactive oxygen species production and Ca(2+) overload. Biochem. Biophys. Res. Commun. 2010, 399, 167–172. [Google Scholar] [CrossRef]
- Guan, X.H.; Liu, X.H.; Hong, X.; Zhao, N.; Xiao, Y.F.; Wang, L.F.; Tang, L.; Jiang, K.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Protects the Heart from Ischemia/Reperfusion Injury through Activating SIRT1/FOXOs-Mediated Antioxidative Stress Pathway. Oxid. Med. Cell Longev. 2016, 2016, 7410257. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Zhang, Q.; Wei, Q.; Lin, J.; Jia, J.; Zhang, J.; Yan, T.; Lv, Y.; Jiang, X.; et al. CD38 Causes Autophagic Flux Inhibition and Cardiac Dysfunction Through a Transcriptional Inhibition Pathway Under Hypoxia/Ischemia Conditions. Front. Cell Dev. Biol. 2020, 8, 191. [Google Scholar] [CrossRef]
- Wang, L.F.; Huang, C.C.; Xiao, Y.F.; Guan, X.H.; Wang, X.N.; Cao, Q.; Liu, Y.; Huang, X.; Deng, L.B.; Deng, K.Y.; et al. CD38 Deficiency Protects Heart from High Fat Diet-Induced Oxidative Stress Via Activating Sirt3/FOXO3 Pathway. Cell Physiol. Biochem. 2018, 48, 2350–2363. [Google Scholar] [CrossRef]
- Slivnick, J.; Lampert, B.C. Hypertension and Heart Failure. Heart Fail. Clin. 2019, 15, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Guan, X.H.; Hong, X.; Zhao, N.; Liu, X.H.; Xiao, Y.F.; Chen, T.T.; Deng, L.B.; Wang, X.L.; Wang, J.B.; Ji, G.J.; et al. CD38 promotes angiotensin II-induced cardiac hypertrophy. J. Cell Mol. Med. 2017, 21, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Gan, L.; Jiang, W.; Xiao, Y.F.; Deng, L.; Gu, L.D.; Guo, Z.Y.; Zhou, Z.C.; Wu, D.; Xin, H.B. Disruption of CD38 gene enhances cardiac functions by elevating serum testosterone in the male null mice. Life Sci. 2011, 89, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Agorrody, G.; Peclat, T.R.; Peluso, G.; Gonano, L.A.; Santos, L.; van Schooten, W.; Chini, C.C.S.; Escande, C.; Chini, E.N.; Contreras, P. Benefits in cardiac function by CD38 suppression: Improvement in NAD(+) levels, exercise capacity, heart rate variability and protection against catecholamine-induced ventricular arrhythmias. J. Mol. Cell Cardiol. 2022, 166, 11–22. [Google Scholar] [CrossRef]
- de Zélicourt, A.; Fayssoil, A.; Dakouane-Giudicelli, M.; De Jesus, I.; Karoui, A.; Zarrouki, F.; Lefebvre, F.; Mansart, A.; Launay, J.M.; Piquereau, J.; et al. CD38-NADase is a new major contributor to Duchenne muscular dystrophic phenotype. EMBO. Mol. Med. 2022, 14, e12860. [Google Scholar] [CrossRef]
- Gan, L.; Liu, D.; Liu, J.; Chen, E.; Chen, C.; Liu, L.; Hu, H.; Guan, X.; Ma, W.; Zhang, Y.; et al. CD38 deficiency alleviates Ang II-induced vascular remodeling by inhibiting small extracellular vesicle-mediated vascular smooth muscle cell senescence in mice. Signal. Transduct. Target. Ther. 2021, 6, 223. [Google Scholar] [CrossRef]
- Reyes, L.A.; Boslett, J.; Varadharaj, S.; De Pascali, F.; Hemann, C.; Druhan, L.J.; Ambrosio, G.; El-Mahdy, M.; Zweier, J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. USA 2015, 112, 11648–11653. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Zhao, Y.J.; Lee, H.C.; Zweier, J.L. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD(P)(H). J. Pharmacol. Exp. Ther. 2017, 361, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Boslett, J.; Hemann, C.; Christofi, F.L.; Zweier, J.L. Characterization of CD38 in the major cell types of the heart: Endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am. J. Physiol. Cell Physiol. 2018, 314, C297–C309. [Google Scholar] [CrossRef] [PubMed]
- Boslett, J.; Helal, M.; Chini, E.; Zweier, J.L. Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. J. Mol. Cell Cardiol. 2018, 118, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Wei, W.; Li, X.X.; Xu, M. Regulation of NLRP3 inflammasome by CD38 through cADPR-mediated Ca(2+) release in vascular smooth muscle cells in diabetic mice. Life Sci. 2020, 255, 117758. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.X.; Zhang, Q.F.; Wang, M.; Xia, M.; Boini, K.M.; Gulbins, E.; Zhang, Y.; Li, P.L. Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front. Biosci. 2017, 22, 558–569. [Google Scholar] [CrossRef]
- Yuan, X.; Bhat, O.M.; Meng, N.; Lohner, H.; Li, P.L. Protective Role of Autophagy in Nlrp3 Inflammasome Activation and Medial Thickening of Mouse Coronary Arteries. Am. J. Pathol. 2018, 188, 2948–2959. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, X.; Li, N.; Dewey, W.L.; Li, P.L.; Zhang, F. Lysosomal cholesterol accumulation in macrophages leading to coronary atherosclerosis in CD38(−/−) mice. J. Cell Mol. Med. 2016, 20, 1001–1013. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.X.; Wang, L.; Wang, M.; Zhang, Y.; Li, P.L. Contribution of Nrf2 to Atherogenic Phenotype Switching of Coronary Arterial Smooth Muscle Cells Lacking CD38 Gene. Cell Physiol. Biochem. 2015, 37, 432–444. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Thai, T.L.; Arendshorst, W.J. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am. J. Physiol. Renal. Physiol. 2009, 297, F169–F176. [Google Scholar] [CrossRef]
- Mitsui-Saito, M.; Kato, I.; Takasawa, S.; Okamoto, H.; Yanagisawa, T. CD38 gene disruption inhibits the contraction induced by alpha-adrenoceptor stimulation in mouse aorta. J. Vet. Med. Sci. 2003, 65, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Zhang, Y.; Xia, M.; Li, X.X.; Ritter, J.K.; Zhang, F.; Li, P.L. NAD(P)H oxidase-dependent intracellular and extracellular O2•- production in coronary arterial myocytes from CD38 knockout mice. Free Radic. Biol. Med. 2012, 52, 357–365. [Google Scholar] [CrossRef]
- Moss, N.G.; Vogel, P.A.; Kopple, T.E.; Arendshorst, W.J. Thromboxane-induced renal vasoconstriction is mediated by the ADP-ribosyl cyclase CD38 and superoxide anion. Am. J. Physiol. Renal Physiol. 2013, 305, F830–F838. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef]
- Boini, K.M.; Xia, M.; Xiong, J.; Li, C.; Payne, L.P.; Li, P.L. Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis. J. Cell Mol. Med. 2012, 16, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Xia, M.; Xu, M.; Zhang, Y.; Abais, J.M.; Li, G.; Riebling, C.R.; Ritter, J.K.; Boini, K.M.; Li, P.L. Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J. Cell Mol. Med. 2013, 17, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Yokota, L.G.; Sampaio, B.M.; Rocha, E.P.; Balbi, A.L.; Sousa Prado, I.R.; Ponce, D. Acute kidney injury in elderly patients: Narrative review on incidence, risk factors, and mortality. Int. J. Nephrol. Renovasc. Dis. 2018, 11, 217–224. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Li, Q.; Wu, C.; Liu, Z.; Zhang, H.; Du, Y.; Liu, Y.; Song, K.; Shi, Q.; Li, R. Increased TLR4 Expression Aggravates Sepsis by Promoting IFN-γ Expression in CD38(−/−) Mice. J. Immunol. Res. 2019, 2019, 3737890. [Google Scholar] [CrossRef]
- van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Malavasi, F.; Faini, A.C.; Morandi, F.; Castella, B.; Incarnato, D.; Oliviero, S.; Horenstein, A.L.; Massaia, M.; van de Donk, N.; Richardson, P.G. Molecular dynamics of targeting CD38 in multiple myeloma. Br. J. Haematol. 2021, 193, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Corzo, K.; Chiron, M.; Velde, H.V.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells. 2019, 8, 1522. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Xiao, S.; Chen, H.; Zhang, M.; Chen, Z.; Long, Y.; Gao, L.; Zhu, G.; He, J.; Peng, S.; et al. CD38 enhances the proliferation and inhibits the apoptosis of cervical cancer cells by affecting the mitochondria functions. Mol. Carcinog. 2017, 56, 2245–2257. [Google Scholar] [CrossRef]
- Chmielewski, J.P.; Bowlby, S.C.; Wheeler, F.B.; Shi, L.; Sui, G.; Davis, A.L.; Howard, T.D.; D’Agostino, R.B., Jr.; Miller, L.D.; Sirintrapun, S.J.; et al. CD38 Inhibits Prostate Cancer Metabolism and Proliferation by Reducing Cellular NAD(+) Pools. Mol. Cancer Res. 2018, 16, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.; Guerrico, A.M.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer cells: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Du, X.; Li, J.; Qin, F.X. Evolving roles of CD38 metabolism in solid tumour microenvironment. Br. J. Cancer 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Protective Effect of CD38 Deficiency/Inhibition against Heart Injury | |||
|---|---|---|---|
| Experimental Model | Animal/Cells | Effect | Reference |
| Ischemia/reperfusion Hypoxia/reoxygenation | CD38KO mice (C57BL/6 mice) CD38 knockdown H9c2 cells | Infarct size↓ Sirt1↑ FoxO1,3↑ SOD2↑ catalase↑ Nox4↓ Ca2+↓ | [108] |
| Hypoxia/ischemic thermal burn model | CD38KO mice (B2.129P2-CD38tm1Lnd backcrossed to a C57BL/6J for more than 10 generations) Cultured neonatal mouse cardiomyocytes | PLEKHM1↑ Sirt1↑ FoxO1↑ Rab7↑ Autophagy↑ | [109] |
| High-fat diet | CD38KO mice (C57BL/6 mice) CD38 knockdown H9c2 cells with oleic acid | GSH↓ FFA↓ NAD+↑ ROS↓lipid synthesis↓ Sirt3↑FoxO3↑SOD2↑ Nox2↓Nox4↓ | [110] |
| Ang-II infusion | CD38KO mice (C57BL/6 mice) CD38 knockdown H9c2 cells | Cardiac hypertrophy↓ Sirt3↑AMPK↑ FoxO3↑ SOD2↑catalase↑ ERK↓Akt↓GSK3β↓ Ca2+↓NFATc↓ | [113] |
| d-galactose (d-gal)-induced myocardial cell senescence | CD38 knockdown H9c2 cells CD38 knockdown with Sirt1 specific inhibitor Ex-527 | SA-β-Galactose↓ ROS↓Nox4↓SOD2↑ Autophagy↑ Cellular senescence↑ ROS↑ | [114] |
| Male mice | CD38KO male (C57BL/6 mice) | Serum testosterone↑ RyR2↑ SERCA2↑ NCX1↑α-MHC↑ Myocardial contraction↑ | [115] |
| Caffeine–epinephrine stimulation | CD38KO mice (C57BL/6 mice) Genetically modified mice with catalytically inactive of CD38 WT mice treated with antibody 68 | Exercise capacity↑ Basal heart rates↓ Heart rate variability↑ SERCA2a↑ Frequency of spontaneous Ca2+ release under stressful conditions↓ Ventricular tachycardia↓ | [116] |
| Isoproterenol stimulation | CD38KO mice (C57BL/6 mice) | Arrhythmogenicity↓ | [55] |
| Duchenne muscular dystrophy model, isoproterenol stimulation | mdx/CD38KO mice | Cardiac function↑ Cardiac fibrosis↓ Mortality due to heart failure↓ hypertrophy↓ Ca2+ sparks and waves in cardiomyocytes at rest↓ | [117] |
| CD38 Deficiency/Inhibition Protects Vascular Tissue And Cells | |||
|---|---|---|---|
| Experimental Model | Animal/Vascular Tissue/Cells | Effect | Reference |
| Ang-II-induced hypertension | CD38KO mice (C57BL/6 mice) Cultured mice VSMCs | Hypertension↓ Vascular remodeling↓ VSMCs senescence↓ Sirt1↑Mitophagy↑ Sirt3↑SA-sEVs biosynthesis/secretion↓ | [118] |
| Ischemia/reperfusion | CD38 knockdown or CD38 inhibitor α-NAD in cultured RAECs C57BL/6J mice treated with α-NAD | NADP(H)↑ NO↑O2−↓BH4↑ Recovery of contractile function↑ Infarct size of the LV↓ | [119] |
| Hypoxia/reoxygenation | RAECs with CD38 knockdown | NADP(H)↑ NO↑O2−↓ | [121] |
| Ischemia/reperfusion | CD38KO mice (C57BL/6 mice) | NADP(H)↑ NO↑O2−↓BH4↑GSH↑ Recovery of contractile function↑ Infarct size of the LV↓ | [122] |
| Ischemia/reperfusion | Sprague Dawley rats treated with luteolinidin | NADP(H)↑ NO↑O2−↓BH4↑ Recovery of contractile function↑ Coronary flow↑ Infarct size of the LV↓ | [120] |
| High-glucose condition | Mice VSMCs with CD38 knockdown | Inflammasome↓ VSMCs proliferation↓ Collagen I synthesis↓ | [123] |
| CD38 deficiency/inhibition promotes coronary atherosclerosis | |||
| High-fat diet | CD38KO mice (C57BL/6 mice) Coronary artery myocytes from CD38KO mice | Coronary artery collagen I deposition↑ Autophagy↓Collagen I degradation↓/accumulation↑ | [124] |
| High-fat diet | CD38KO mice (C57BL/6 mice) coronary artery Coronary artery SMCs from CD38KO mice | Autophagy↓ Inflammasome↑ Inflammation↑ Remodeling↑ | [125] |
| High-fat diet | CD38KO mice (C57BL/6 mice) coronary artery Bone-marrow-derived macrophages from CD38KO mice | Intimal and media layer thickening↑ Aggregation of macrophages↑ Deposition of free cholesterol↑ NAADP induced Ca2+↓ Free cholesterol efflux from lysosome↓ | [126] |
| 7-ketocholesterol stimulation | CAMs from CD38KO mice (C57BL/6 mice) | cADPR/NAADP induced Ca2+↓ O2− via Nox4↓ Nrf2↓ SMCs proliferative and atherosclerotic phenotype↓ | [127] |
| The Protective Effect of CD38 Deficiency/Inhibition against Kidney Injury | |||
|---|---|---|---|
| Experimental Model | Animal/Cells | Effect | Reference |
| Diabetic kidney disease High-glucose condition | Zucker diabetic fatty rats treated with apigenin HK-2 cells treated with apigenin | NAD+/NADH↑ Sirt3↑ Acetylated-SOD2↓Acetylated-IDH2↓ Mitochondrial ROS↓ Tubulo-interstitial inflammation↓fibrosis↓ | [133] |
| LPS-induced AKI | C57BL/6 mice treated with quercetin CD38 knockdown quercetin RAW 264.7 cells | Tubular injury↓ Inflammation↓ Serum Urea↓ Macrophage M1 polarization↓ NF-κB↓inflammation↓ | [74] |
| CD38 deficiency or inhibition in promoting kidney injury | |||
| Doca+1% NaCl treatment | CD38KO mice (C57BL/6 mice) C57BL/6J mice with CD38 shRNA Conditionally immortalized mouse podocytes with CD38 shRNA | Podocytes EMT↑ Glomerular injury↑ Blood pressure↑ Urinary protein/albumin excretion↑ | [134] |
| Cell culture | Conditionally immortalized mouse podocytes with CD38 shRNA | NAADP-mediated Ca2+ release↓ Lysosome fusion to autophagosome↓ | [135] |
| LPS-induced AKI | CD38−/− mice (B6.129P2-Ly96−/−) | Serum Cr↑ Tubular injury↑ Inflammation↑ NF-κB↑ TRL4↑IFN-γ↑ | [138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitada, M.; Araki, S.-i.; Koya, D. The Role of CD38 in the Pathogenesis of Cardiorenal Metabolic Disease and Aging, an Approach from Basic Research. Cells 2023, 12, 595. https://doi.org/10.3390/cells12040595
Kitada M, Araki S-i, Koya D. The Role of CD38 in the Pathogenesis of Cardiorenal Metabolic Disease and Aging, an Approach from Basic Research. Cells. 2023; 12(4):595. https://doi.org/10.3390/cells12040595
Chicago/Turabian StyleKitada, Munehiro, Shin-ichi Araki, and Daisuke Koya. 2023. "The Role of CD38 in the Pathogenesis of Cardiorenal Metabolic Disease and Aging, an Approach from Basic Research" Cells 12, no. 4: 595. https://doi.org/10.3390/cells12040595