

MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges


Abstract
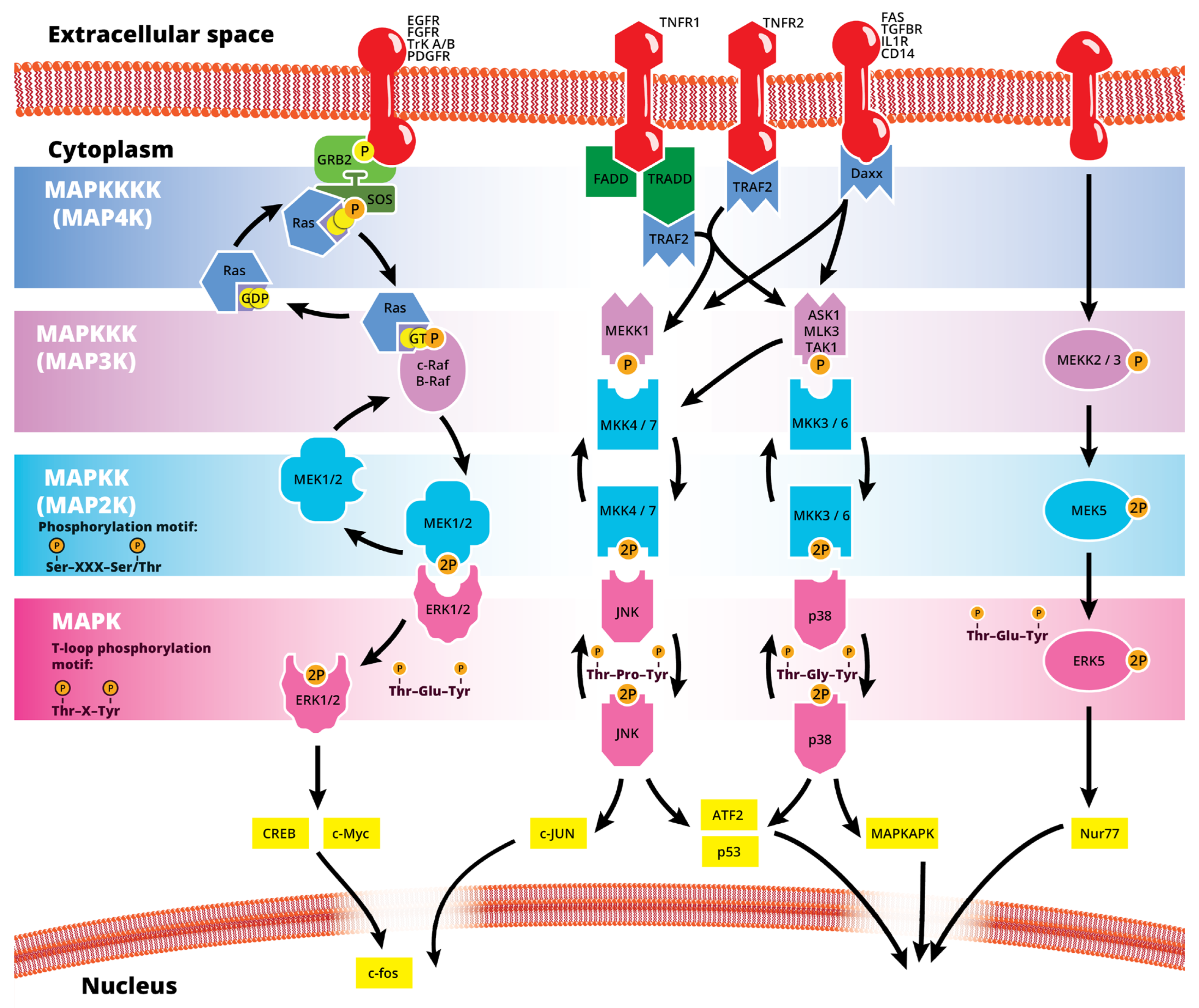
1. Introduction
2. Physiological Role of MAPKs in the Eye
3. Link between the MAPK Pathway and Ocular Pathophysiology
3.1. MAPK in the Retina
3.2. MAPK in the Cornea
3.3. MAPK in Other Ocular Pathophysiologies
3.4. MAPK InhibitorAdverse Effects

{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Kinase Inhibited | Adverse Effects | Frequency |
|---|---|---|---|
| Binimetinib [213,219,227] | MEK | MEKAR | 9–62% |
| Subfoveal neurosensory retinal detachment | |||
| Pigment epithelial detachment | |||
| Retinal vein occlusion | |||
| Dry eye | 10.0% | ||
| Blurred vision | 20.0% | ||
| Visual impairment | |||
| Retinal thinning | |||
| Visual symptoms | |||
| Pimasertib [213,222,227,228] | MEK | MEKAR | 33–50% |
| Serous retinal detachment | 10.0% | ||
| Macular degeneration | 24% | ||
| Visual field defects | |||
| Macular edema | |||
| Retinal vein occlusion | |||
| Blurred vision | 17.0% | ||
| Visual impairment | |||
| Cobimetinib [213,223] | MEK | MEKAR | 11.7% |
| Floaters | |||
| Periorbital edema | |||
| Ocular icterus | |||
| Blurred vision | 4.75–50% | ||
| Visual impairment | |||
| Increased lacrimation | |||
| Photophobia | |||
| Trametinib [213] | MEK | MEKAR | 1–2% |
| Retinal vein occlusion | |||
| Dry eye | 7% | ||
| Eye pain | 5.5% | ||
| Increased IOP | |||
| Conjunctivitis | |||
| Selumetinib [213,229] | MEK | MEKAR | 1.0% |
| Retinal vein occlusion | 1.0% | ||
| Ocular hypertension | 15% | ||
| Floaters | |||
| Epiphora | |||
| Periorbital edema | |||
| Blurred vision | 30.0% | ||
| Light disturbances | |||
| RPE detachment | |||
| Refametinib [213] | MEK | MEKAR | |
| Retinal vein occlusion | |||
| Floaters | |||
| Cataracts | |||
| Dry eye | |||
| Blurred vision | |||
| Ulixertinib [224] | ERK | MEKAR | 80% |
| Multifocal subretinal fluid | |||
| Blurry vision | 20.0% | ||
| KO-947 [224] | ERK | Multifocal subretinal fluid | |
| Blurry vision | 11.0% | ||
| Metamorphopsia | 44.0% | ||
| Dabrafenib [227,230,231] | b-Raf | Uveitis | 1–10% |
| Blurred vision | 1–10% | ||
| Eye pain | |||
| Change in color vision | |||
| Photophobia | |||
| Eye redness | |||
| Tearing |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef]
- Gkouveris, I.; Nikitakis, N.G. Role of JNK signaling in oral cancer: A mini review. Tumor Biol. 2017, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Büscher, D.; Hipskind, R.A.; Krautwald, S.; Reimann, T.; Baccarini, M. Ras-dependent and -independent pathways target the mitogen-activated protein kinase network in macrophages. Mol. Cell. Biol. 1995, 15, 466–475. [Google Scholar] [CrossRef]
- Brown, J.L.; Sones, J.L.; Angulo, C.N.; Abbott, K.; Miller, A.D.; Boehm, U.; Roberson, M.S. Conditional loss of ERK1 and ERK2 results in abnormal placentation and delayed parturition in the mouse. Sci. Rep. 2019, 9, 9641. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, T.-T.; Li, X.; Huang, X.-Q.; Chen, X.-W.; Li, Z.-L.; Lv, X.-M.; Liu, F.-Y.; Luo, Z.-W.; et al. ERK Signaling Pathway Regulates Embryonic Survival and Eye Development in Goldfish, Carassius auratus. Curr. Mol. Med. 2013, 13, 959–967. [Google Scholar] [CrossRef]
- Jafry, M.; Sidbury, R. RASopathies. Clin. Dermatol. 2020, 38, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Alrejaye, N.; Klein, O.D.; Goodwin, A.F.; Oberoi, S. A review of craniofacial and dental findings of the RASopathies. Orthod. Craniofac. Res. 2017, 20 (Suppl. S1), 32–38. [Google Scholar] [CrossRef]
- Hecquet, C.; Lefevre, G.; Valtink, M.; Engelmann, K.; Mascarelli, F. Activation and role of MAP kinase-dependent pathways in retinal pigment epithelial cells: ERK and RPE cell proliferation. Investig. Opthalmol. Vis. Sci. 2002, 43, 3091–3098. [Google Scholar]
- Yasumuro, H.; Sakurai, K.; Toyama, F.; Maruo, F.; Chiba, C. Implications of a Multi-Step Trigger of Retinal Regeneration in the Adult Newt. Biomedicines 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Kochańczyk, M.; Kocieniewski, P.; Kozłowska, E.; Jaruszewicz-Błońska, J.; Sparta, B.; Pargett, M.; Albeck, J.G.; Hlavacek, W.S.; Lipniacki, T. Relaxation oscillations and hierarchy of feedbacks in MAPK signaling. Sci. Rep. 2017, 7, 38244. [Google Scholar] [CrossRef]
- Mizuno, A.; Yasumuro, H.; Yoshikawa, T.; Inami, W.; Chiba, C. MEK–ERK signaling in adult newt retinal pigment epithelium cells is strengthened immediately after surgical induction of retinal regeneration. Neurosci. Lett. 2012, 523, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Mizuno, A.; Yasumuro, H.; Inami, W.; Vergara, M.N.; Del Rio-Tsonis, K.; Chiba, C. MEK-ERK and heparin-susceptible signaling pathways are involved in cell-cycle entry of the wound edge retinal pigment epithelium cells in the adult newt. Pigment Cell Melanoma Res. 2012, 25, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Susaki, K.; Chiba, C. MEK mediates in vitro neural transdifferentiation of the adult newt retinal pigment epithelium cells: Is FGF2 an induction factor? Pigment Cell Res. 2007, 20, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Vergara, M.N.; Del Rio-Tsonis, K. Retinal regeneration in the Xenopus laevis tadpole: A new model system. Mol. Vis. 2009, 15, 1000–1013. [Google Scholar] [PubMed]
- Wan, J.; Zhao, X.-F.; Vojtek, A.; Goldman, D. Retinal injury, growth factors, and cytokines converge on β-catenin and pStat3 signaling to stimulate retina regeneration. Cell Rep. 2014, 9, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Ramachandran, R.; Goldman, D. HB-EGF is Necessary and Sufficient for Müller Glia Dedifferentiation and Retina Regeneration. Dev. Cell 2012, 22, 334–347. [Google Scholar] [CrossRef]
- Naska, S.; Cenni, M.C.; Menna, E.; Maffei, L. ERK signaling is required for eye-specific retino-geniculate segregation. Development 2004, 131, 3559–3570. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Madhavan, M.; Aycinena, J.-C.; Del Rio-Tsonis, K. Retina regeneration in the chick embryo is not induced by spontaneous Mitf downregulation but requires FGF/FGFR/MEK/Erk dependent upregulation of Pax6. Mol. Vis. 2007, 13, 57–65. [Google Scholar]
- Galy, A.; Néron, B.; Planque, N.; Saule, S.; Eychène, A. Activated MAPK/ERK kinase (MEK-1) induces transdifferentiation of pigmented epithelium into neural retina. Dev. Biol. 2002, 248, 251–264. [Google Scholar] [CrossRef]
- Fischer, A.J.; Scott, M.A.; Tuten, W. Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia 2009, 57, 166–181. [Google Scholar] [CrossRef]
- Syc-Mazurek, S.B.; Rausch, R.L.; Fernandes, K.A.; Wilson, M.P.; Libby, R.T. Mkk4 and Mkk7 are important for retinal development and axonal injury-induced retinal ganglion cell death. Cell Death Dis. 2018, 9, 1095. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.; Shaikh, A.R.; Hennelly, M.M.; Li, Q.; Bindokas, V.; Graham, C.E. Mitogen-Activated Protein Kinases and Retinal is chemia. Investig. Opthalmol. Vis. Sci. 2003, 44, 5383–5395. [Google Scholar] [CrossRef]
- Agca, C.; Gubler, A.; Traber, G.; Beck, C.; Imsand, C.; Ail, D.; Caprara, C.; Grimm, C. p38 MAPK signaling acts upstream of LIF-dependent neuroprotection during photoreceptor degeneration. Cell Death Dis. 2013, 4, e785. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, M.; Tenneti, L.; Lipton, S.A. Role of p38 Mitogen-Activated Protein Kinase in Axotomy-Induced Apoptosis of Rat Retinal Ganglion Cells. J. Neurosci. 2000, 20, 5037–5044. [Google Scholar] [CrossRef]
- Zhang, S.S.-M.; Li, H.; Huang, P.; Lou, L.X.; Fu, X.-Y.; Barnstable, C.J. MAPK signaling during Müller glial cell development in retina explant cultures. J. Ocul. Biol. Dis. Inform. 2010, 3, 129–133. [Google Scholar] [CrossRef]
- Cai, Z.; Simons, D.L.; Fu, X.-Y.; Feng, G.-S.; Wu, S.M.; Zhang, X. Loss of Shp2-Mediated Mitogen-Activated Protein Kinase Signaling in Müller Glial Cells Results in Retinal Degeneration. Mol. Cell. Biol. 2011, 31, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.S.; Holt, C.E. Apoptotic Pathway and MAPKs Differentially Regulate Chemotropic Responses of Retinal Growth Cones. Neuron 2003, 37, 939–952. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Lee, P.H. AMD-Associated Genes Encoding Stress-Activated MAPK Pathway Constituents Are Identified by Interval-Based Enrichment Analysis. PLoS ONE 2013, 8, e71239. [Google Scholar] [CrossRef] [PubMed]
- Makarev, E.; Cantor, C.; Zhavoronkov, A.; Buzdin, A.; Aliper, A.; Csoka, A.B. Pathway activation profiling reveals new insights into Age-related Macular Degeneration and provides avenues for therapeutic interventions. Aging 2014, 6, 1064–1075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Gil Cho, W.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Dridi, S.; Hirano, Y.; Tarallo, V.; Kim, Y.; Fowler, B.J.; Ambati, B.K.; Bogdanovich, S.; Chiodo, V.A.; Hauswirth, W.W.; Kugel, J.F.; et al. ERK1/2 activation is a therapeutic target in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 13781–13786. [Google Scholar] [CrossRef]
- Kyosseva, S.V.; Chen, L.; Seal, S.; McGinnis, J.F. Nanoceria inhibit expression of genes associated with inflammation and angiogenesis in the retina of Vldlr null mice. Exp. Eye Res. 2013, 116, 63–74. [Google Scholar] [CrossRef]
- Du, H.; Sun, X.; Guma, M.; Luo, J.; Ouyang, H.; Zhang, X.; Zeng, J.; Quach, J.; Nguyen, D.H.; Shaw, P.X.; et al. JNK inhibition reduces apoptosis and neovascularization in a murine model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 2377–2382. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870. [Google Scholar] [CrossRef]
- Roduit, R.; Schorderet, D. MAP kinase pathways in UV-induced apoptosis of retinal pigment epithelium ARPE19 cells. Apoptosis 2008, 13, 343–353. [Google Scholar] [CrossRef]
- Chan, C.-M.; Huang, J.-H.; Lin, H.-H.; Chiang, H.-S.; Chen, B.-H.; Hong, J.-Y.; Hung, C.-F. Protective effects of (-)-epigallocatechin gallate on UVA-induced damage in ARPE19 cells. Mol. Vis. 2008, 14, 2528–2534. [Google Scholar]
- Cao, G.; Chen, M.; Song, Q.; Liu, Y.; Xie, L.; Han, Y.; Liu, Z.; Ji, Y.; Jiang, Q. EGCG protects against UVB-induced apoptosis via oxidative stress and the JNK1/c-Jun pathway in ARPE19 cells. Mol. Med. Rep. 2012, 5, 54–59. [Google Scholar]
- Chu, Y.K.; Lee, S.C.; Byeon, S.H. VEGF Rescues Cigarette Smoking–Induced Human RPE Cell Death by Increasing Autophagic Flux: Implications of the Role of Autophagy in Advanced Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2013, 54, 7329–7337. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A., Jr.; Grant, M.B.; Boulton, M.E. Autophagy in the Retina: A Potential Role in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2012, 723, 83–90. [Google Scholar] [CrossRef]
- Glotin, A.L.; Calipel, A.; Brossas, J.Y.; Faussat, A.M.; Tréton, J.; Mascarelli, F. Sustained versus transient ERK1/2 signaling underlies the anti- and proapoptotic effects of oxidative stress in human RPE cells. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4614–4623. [Google Scholar] [CrossRef] [PubMed]
- Pyakurel, A.; Balmer, D.; Saba-El-Leil, M.K.; Kizilyaprak, C.; Daraspe, J.; Humbel, B.M.; Voisin, L.; Le, Y.Z.; von Lintig, J.; Meloche, S.; et al. Loss of Extracellular Signal-Regulated Kinase 1/2 in the Retinal Pigment Epithelium Leads to RPE65 Decrease and Retinal Degeneration. Mol. Cell. Biol. 2017, 37, e00295-17. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; McLeod, D.S.; Hasegawa, T.; Kim, S.Y.; Merges, C.; Tong, P.; Lutty, G.A. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp. Eye Res. 2006, 82, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Ablonczy, Z.; Dahrouj, M.; Marneros, A.G. Progressive dysfunction of the retinal pigment epithelium and retina due to increased VEGF-A levels. FASEB J. 2014, 28, 2369–2379. [Google Scholar] [CrossRef]
- Luo, X.; Gu, S.; Zhang, Y.; Zhang, J. Kinsenoside Ameliorates Oxidative Stress-Induced RPE Cell Apoptosis and Inhibits Angiogenesis via Erk/p38/NF-κB/VEGF Signaling. Front. Pharmacol. 2018, 9, 240. [Google Scholar] [CrossRef]
- Klettner, A.; Roider, J. Constitutive and oxidative-stress-induced expression of VEGF in the RPE are differently regulated by different Mitogen-activated protein kinases. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 1487–1492. [Google Scholar] [CrossRef]
- Koinzer, S.; Reinecke, K.; Herdegen, T.; Roider, J.; Klettner, A. Oxidative Stress Induces Biphasic ERK1/2 Activation in the RPE with Distinct Effects on Cell Survival at Early and Late Activation. Curr. Eye Res. 2015, 40, 853–857. [Google Scholar] [CrossRef]
- Milanini, J.; Viñals, F.; Pouysségur, J.; Pagès, G. p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J. Biol. Chem. 1998, 273, 18165–18172. [Google Scholar] [CrossRef]
- Pagès, G.; Berra, E.; Milanini, J.; Levy, A.P.; Pouysségur, J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J. Biol. Chem. 2000, 275, 26484–26491. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, T.; DU, S.; Chen, Y.; Wang, S.; Xiong, F.; Wu, Q. The MAPK signaling pathway mediates the GPR91-dependent release of VEGF from RGC-5 cells. Int. J. Mol. Med. 2015, 36, 130–138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klettner, A. Oxidative stress induced cellular signaling in RPE cells. Front. Biosci. (Schol. Ed.) 2012, 4, 392–411. [Google Scholar] [CrossRef] [PubMed]
- Courtaut, F.; Scagliarini, A.; Aires, V.; Cornebise, C.; de Barros, J.P.P.; Olmiere, C.; Delmas, D. VEGF-R2/Caveolin-1 Pathway of Undifferentiated ARPE-19 Retina Cells: A Potential Target as Anti-VEGF-A Therapy in Wet AMD by Resvega, an Omega-3/Polyphenol Combination. Int. J. Mol. Sci. 2021, 22, 6590. [Google Scholar] [CrossRef] [PubMed]
- Sghaier, R.; Perus, M.; Cornebise, C.; Courtaut, F.; Scagliarini, A.; Olmiere, C.; Aires, V.; Hermetet, F.; Delmas, D. Resvega, a Nutraceutical Preparation, Affects NFκB Pathway and Prolongs the Anti-VEGF Effect of Bevacizumab in Undifferentiated ARPE-19 Retina Cells. Int. J. Mol. Sci. 2022, 23, 11704. [Google Scholar] [CrossRef] [PubMed]
- Courtaut, F.; Aires, V.; Acar, N.; Bretillon, L.; Guerrera, I.C.; Chhuon, C.; de Barros, J.-P.P.; Olmiere, C.; Delmas, D. RESVEGA, a Nutraceutical Omega-3/Resveratrol Supplementation, Reduces Angiogenesis in a Preclinical Mouse Model of Choroidal Neovascularization. Int. J. Mol. Sci. 2021, 22, 11023. [Google Scholar] [CrossRef]
- Bielmeier, C.B.; Schmitt, S.I.; Kleefeldt, N.; Boneva, S.K.; Schlecht, A.; Vallon, M.; Tamm, E.R.; Hillenkamp, J.; Ergün, S.; Neueder, A.; et al. Deficiency in Retinal TGFβ Signaling Aggravates Neurodegeneration by Modulating Pro-Apoptotic and MAP Kinase Pathways. Int. J. Mol. Sci. 2022, 23, 2626. [Google Scholar] [CrossRef]
- Tsao, Y.-P.; Ho, T.-C.; Chen, S.-L.; Cheng, H.-C. Pigment epithelium-derived factor inhibits oxidative stress-induced cell death by activation of extracellular signal-regulated kinases in cultured retinal pigment epithelial cells. Life Sci. 2006, 79, 545–550. [Google Scholar] [CrossRef]
- Ye, M.-J.; Meng, N. Resveratrol acts via the mitogen-activated protein kinase (MAPK) pathway to protect retinal ganglion cells from apoptosis induced by hydrogen peroxide. Bioengineered 2021, 12, 4878–4886. [Google Scholar] [CrossRef]
- Ho, T.-C.; Yang, Y.-C.; Cheng, H.-C.; Wu, A.-C.; Chen, S.-L.; Chen, H.-K.; Tsao, Y.-P. Activation of mitogen-activated protein kinases is essential for hydrogen peroxide -induced apoptosis in retinal pigment epithelial cells. Apoptosis 2006, 11, 1899–1908. [Google Scholar] [CrossRef]
- Kyosseva, S.V. Targeting MAPK Signaling in Age-Related Macular Degeneration. Ophthalmol. Eye Dis. 2016, 8, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent Activation of ERK Contributes to Glutamate-induced Oxidative Toxicity in a Neuronal Cell Line and Primary Cortical Neuron Cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef]
- Schur, R.M.; Gao, S.; Yu, G.; Chen, Y.; Maeda, A.; Palczewski, K.; Lu, Z.-R. New GABA modulators protect photoreceptor cells from light-induced degeneration in mouse models. FASEB J. 2018, 32, 3289–3300. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Y.; Gu, R.P.; Tang, W.Y.; Shu, Q.M.; Xu, G.Z.; Zhang, M. Effect of Phosphorylated-Extracellular Regulated Kinase 1/2 Inhibitor on Retina from Light-induced Photoreceptor Degeneration. Chin. Med. J. 2018, 131, 2836–2843. [Google Scholar] [PubMed]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Knudtson, M.D.; Klein, B.E.; Wong, T.Y.; Cotch, M.F.; Liu, K.; Cheng, C.Y.; Burke, G.L.; Saad, M.F.; Jacobs, D.R.; et al. Inflammation, Complement Factor H, and Age-Related Macular Degeneration: The Multi-Ethnic Study of Atherosclerosis. Ophthalmology 2008, 115, 1742–1749. [Google Scholar] [CrossRef]
- Qin, T.; Gao, S. Inhibition of Proteasome Activity Upregulates IL-6 Expression in RPE Cells through the Activation of P38 MAPKs. J. Ophthalmol. 2018, 2018, 5392432. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Bai, Y.; Xie, W.; Shi, X.; Li, F.; Yang, F.; Sun, Y.; Huang, L.; Li, X. Interleukin-1β Level Is Increased in Vitreous of Patients with Neovascular Age-Related Macular Degeneration (nAMD) and Polypoidal Choroidal Vasculopathy (PCV). PLoS ONE 2015, 10, e0125150. [Google Scholar] [CrossRef]
- Cheng, S.C.; Huang, W.C.; JH, S.P.; Wu, Y.H.; Cheng, C.Y. Quercetin Inhibits the Production of IL-1β-Induced Inflammatory Cytokines and Chemokines in ARPE-19 Cells via the MAPK and NF-κB Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 2957. [Google Scholar] [CrossRef]
- Igarashi, M.; Wakasaki, H.; Takahara, N.; Ishii, H.; Jiang, Z.-Y.; Yamauchi, T.; Kuboki, K.; Meier, M.; Rhodes, C.J.; King, G.L. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J. Clin. Investig. 1999, 103, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Tang, J.; Li, G.; Berti-Mattera, L.; Lee, C.A.; Bartkowski, D.; Gale, D.; Monahan, J.; Niesman, M.R.; Alton, G.; et al. Effects of p38 MAPK Inhibition on Early Stages of Diabetic Retinopathy and Sensory Nerve Function. Investig. Opthalmol. Vis. Sci. 2010, 51, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Y.-X. LncRNA HOTTIP improves diabetic retinopathy by regulating the p38-MAPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2941–2948. [Google Scholar]
- Liu, W.Y.; Tzeng, T.F.; Liu, I.M. Zerumbone, a Bioactive Sesquiterpene, Ameliorates Diabetes-Induced Retinal Microvascular Damage through Inhibition of Phospho-p38 Mitogen-Activated Protein Kinase and Nuclear Factor-κB Pathways. Molecules 2016, 21, 1708. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xu, K.; Liu, W.; Liu, A.; Liang, H.; Li, Q.; Feng, Z.; Yang, Y.; Ding, J.; Zhang, T.; et al. Protective Effect of Raf-1 Kinase Inhibitory Protein on Diabetic Retinal Neurodegeneration through P38-MAPK Pathway. Curr. Eye Res. 2022, 47, 135–142. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, H.-J.; Li, H.; Li, Q.-X.; Li, F.-M.; Cheng, L.; Liu, Y.-G. p38-MAPK pathway is activated in retinopathy of microvascular disease of STZ-induced diabetic rat model. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5789–5796. [Google Scholar]
- Matteucci, A.; Gaddini, L.; Villa, M.; Varano, M.; Parravano, M.; Monteleone, V.; Cavallo, F.; Leo, L.; Mallozzi, C.; Malchiodi-Albedi, F.; et al. Neuroprotection by rat Müller glia against high glucose-induced neurodegeneration through a mechanism involving ERK1/2 activation. Exp. Eye Res. 2014, 125, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Scott, M.A.; Ritchey, E.R.; Sherwood, P. Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia 2009, 57, 1538–1552. [Google Scholar] [CrossRef]
- Maugeri, G.; Bucolo, C.; Drago, F.; Rossi, S.; Di Rosa, M.; Imbesi, R.; D’Agata, V.; Giunta, S. Attenuation of High Glucose-Induced Damage in RPE Cells through p38 MAPK Signaling Pathway Inhibition. Front. Pharmacol. 2021, 12, 684680. [Google Scholar] [CrossRef]
- Du, Z.-J.; Kamei, M.; Suzuki, M.; Tano, Y.; Wang, B.-R.; Hui, Y.-N. Coordinated Expression of Ets-1, pERK1/2, and VEGF in Retina of Streptozotocin-Induced Diabetic Rats. Ophthalmic Res. 2007, 39, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Xu, G.; Chang, Q.; Fan, J.; Sun, Z.; Qin, Y.; Jiang, A.C. ERK1/2 signaling pathways involved in VEGF release in diabetic rat retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5226–5533. [Google Scholar] [CrossRef]
- Ye, X.; Ren, H.; Zhang, M.; Sun, Z.; Jiang, A.C.; Xu, G. ERK1/2 signaling pathway in the release of VEGF from Müller cells in diabetes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3481–3489. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ren, H.; Jiang, T.; Zhang, T.; Li, G. Effect of diabetes blood-stasis syndrome and Xuefu Zhuyu decoction on ERK1/2-VEGF signal pathway in rat retina Müller cells. Histol. Histopathol. 2022, 37, 757–767. [Google Scholar] [PubMed]
- Ye, X.; Ren, H.; Jiang, T.; Zhang, T.; Li, G. Effect of diabetes blood-stasis syndrome and Xuefu Zhuyu decoction on ROS-ERK1/2 signaling pathway in rat retina Müller cells. Cytotechnology 2020, 72, 303–314. [Google Scholar] [CrossRef]
- Gregory-Evans, K.; Pennesi, M.E.; Weleber, R.G. Chapter 40—Retinitis Pigmentosa and Allied Disorders. In Retina, 5th ed.; Ryan, S.J., Sadda, S.R., Hinton, D.R., Schachat, A.P., Sadda, S.R., Wilkinson, C.P., Wiedemann, P., Schachat, A.P., Eds.; W.B. Saunders: London, UK, 2013; pp. 761–835. [Google Scholar]
- Wang, A.L.; Knight, D.K.; Vu, T.-T.T.; Mehta, M.C. Retinitis Pigmentosa: Review of Current Treatment. Int. Ophthalmol. Clin. 2019, 59, 263–280. [Google Scholar] [CrossRef]
- Meunier, I.; Lenaers, G.; Bocquet, B.; Baudoin, C.; Piro-Megy, C.; Cubizolle, A.; Quilès, M.; Jean-Charles, A.; Cohen, S.Y.; Merle, H.; et al. A dominant mutation in MAPKAPK3, an actor of p38 signaling pathway, causes a new retinal dystrophy involving Bruch’s membrane and retinal pigment epithelium. Hum. Mol. Genet. 2016, 25, 916–926. [Google Scholar] [CrossRef]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, C.; Yang, Q.; Xie, H.; Liu, D.; Tian, H.; Lu, L.; Xu, J.Y.; Li, W.; Xu, G.; et al. Melatonin maintains inner blood-retinal barrier via inhibition of p38/TXNIP/NF-κB pathway in diabetic retinopathy. J. Cell. Physiol. 2021, 236, 5848–5864. [Google Scholar] [CrossRef]
- Li, Y.; Bai, Y.-J.; Jiang, Y.-R.; Yu, W.-Z.; Shi, X.; Chen, L.; Feng, J.; Sun, G.-B. Apelin-13 Is an Early Promoter of Cytoskeleton and Tight Junction in Diabetic Macular Edema via PI-3K/Akt and MAPK/Erk Signaling Pathways. BioMed Res. Int. 2018, 2018, 3242574. [Google Scholar] [CrossRef]
- Xie, M.-S.; Zheng, Y.-Z.; Huang, L.-B.; Xu, G.-X. Infliximab relieves blood retinal barrier breakdown through the p38 MAPK pathway in a diabetic rat model. Int. J. Ophthalmol. 2017, 10, 1824–1829. [Google Scholar] [CrossRef]
- Wang, S.; Du, S.; Wu, Q.; Hu, J.; Li, T. Decorin Prevents Retinal Pigment Epithelial Barrier Breakdown Under Diabetic Conditions by Suppressing p38 MAPK Activation. Investig. Opthalmol. Vis. Sci. 2015, 56, 2971–2979. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, J.; Lin, C. Sprouty-related proteins with EVH1 domain (SPRED2) prevents high-glucose induced endothelial–mesenchymal transition and endothelial injury by suppressing MAPK activation. Bioengineered 2022, 13, 13882–13892. [Google Scholar] [CrossRef]
- Groeger, G.; Doonan, F.; Cotter, T.G.; Donovan, M. Reactive oxygen species regulate prosurvival ERK1/2 signaling and bFGF expression in gliosis within the retina. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6645–6654. [Google Scholar] [CrossRef]
- Métrailler, S.; Emery, M.; Schorderet, D.F.; Cottet, S.; Roduit, R. ERK1/2 pathway is activated in degenerated Rpe65-deficient mice. Exp. Eye Res. 2013, 116, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhang, T.; Chen, Y.; Chu-Tan, J.; Jin, K.; Lee, S.R.; Yam, M.X.; Madigan, M.C.; Fernando, N.; Cioanca, A.; et al. Inhibiting the activation of MAPK (ERK1/2) in stressed Müller cells prevents photoreceptor degeneration. Theranostics 2022, 12, 6705–6722. [Google Scholar] [CrossRef]
- Gao, F.; Li, F.; Miao, Y.; Xu, L.-J.; Zhao, Y.; Li, Q.; Zhang, S.-H.; Wu, J.; Sun, X.-H.; Wang, Z. Involvement of the MEK-ERK/p38-CREB/c-fos signaling pathway in Kir channel inhibition-induced rat retinal Müller cell gliosis. Sci. Rep. 2017, 7, 1480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, C.; Zhou, T.; Huang, Z.; Zhou, L.; Liu, X. NGF increases VEGF expression and promotes cell proliferation via ERK1/2 and AKT signaling in Müller cells. Mol. Vis. 2016, 22, 254–263. [Google Scholar] [PubMed]
- Jiang, S.-Y.; Zou, Y.-Y.; Wang, J.-T. p38 mitogen-activated protein kinase–induced nuclear factor kappa-light-chain-enhancer of activated B cell activity is required for neuroprotection in retinal ischemia/reperfusion injury. Mol. Vis. 2012, 18, 2096–2106. [Google Scholar] [PubMed]
- Roth, S.; Li, B.; Rosenbaum, P.S.; Gupta, H.; Goldstein, I.; Maxwell, K.M.; Gidday, J.M. Preconditioning provides complete protection against retinal ischemic injury in rats. Investig. Opthalmol. Vis. Sci. 1998, 39, 777–785. [Google Scholar]
- Gesslein, B.; Håkansson, G.; Carpio, R.; Gustafsson, L.; Perez, M.-T.; Malmsjö, M. Mitogen-activated protein kinases in the porcine retinal arteries and neuroretina following retinal ischemia-reperfusion. Mol. Vis. 2010, 16, 392–407. [Google Scholar] [PubMed]
- Dreixler, J.C.; Bratton, A.; Du, E.; Shaikh, A.R.; Savoie, B.; Alexander, M.; Marcet, M.; Roth, S. Mitogen-activated protein kinase phosphatase-1 (MKP-1) in retinal ischemic preconditioning. Exp. Eye Res. 2011, 93, 340–349. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dreixler, J.C.; Barone, F.C.; Shaikh, A.R.; Du, E.; Roth, S. Mitogen-activated protein kinase p38α and retinal ischemic preconditioning. Exp. Eye Res. 2009, 89, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Munemasa, Y.; Ohtani-Kaneko, R.; Kitaoka, Y.; Kumai, T.; Kitaoka, Y.; Hayashi, Y.; Watanabe, M.; Takeda, H.; Hirata, K.; Ueno, S. Pro-apoptotic role of c-Jun in NMDA-induced neurotoxicity in the rat retina. J. Neurosci. Res. 2006, 83, 907–918. [Google Scholar] [CrossRef]
- Munemasa, Y.; Ohtani-Kaneko, R.; Kitaoka, Y.; Kuribayashi, K.; Isenoumi, K.; Kogo, J.; Yamashita, K.; Kumai, T.; Kobayashi, S.; Hirata, K.; et al. Contribution of mitogen-activated protein kinases to NMDA-induced neurotoxicity in the rat retina. Brain Res. 2005, 1044, 227–240. [Google Scholar] [CrossRef]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Wilson, M.P.; Shrager, P.; Libby, R.T. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol. Neurodegener. 2017, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Kilic, E.; Soliz, J.; Bassetti, C.I.; Gassmann, M.; Hermann, D.M. Erythropoietin protects from axotomy-induced degeneration of retinal ganglion cells by activating ERK-1/-2. FASEB J. 2005, 19, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Tamai, M.; Mori, N. Brain-derived neurotrophic factor prevents axotomized retinal ganglion cell death through MAPK and PI3K signaling pathways. Investig. Opthalmol. Vis. Sci. 2002, 43, 3319–3326. [Google Scholar]
- Zavos, C.; Kountouras, J.; Skoura, L.; Sakkias, G.; Parapanisiou, E. Mitogen-activated protein kinase (MAPK) intracellular signalling in the aqueous humour activated by Helicobacter pylori may have a role in glaucoma. Med. Hypotheses 2007, 68, 928–929. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Peng, F.; Liu, Z.; Li, S.; Li, L.; Qian, X. Mechanobiological responses of astrocytes in optic nerve head due to biaxial stretch. BMC Ophthalmol. 2022, 22, 368. [Google Scholar] [CrossRef] [PubMed]
- Pervan, C.L. Smad-independent TGF-β2 signaling pathways in human trabecular meshwork cells. Exp. Eye Res. 2017, 158, 137–145. [Google Scholar] [CrossRef]
- Callaghan, B.; Lester, K.; Lane, B.; Fan, X.; Goljanek-Whysall, K.; Simpson, D.A.; Sheridan, C.; Willoughby, C.E. Genome-wide transcriptome profiling of human trabecular meshwork cells treated with TGF-β2. Sci. Rep. 2022, 12, 9564. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, K.; Haribalaganesh, R.; Krishnadas, R.; Muthukkaruppan, V.; Willoughby, C.E.; Bharanidharan, D.; Senthilkumari, S. A Comparative Genome-Wide Transcriptome Analysis of Glucocorticoid Responder and Non-Responder Primary Human Trabecular Meshwork Cells. Genes 2022, 13, 882. [Google Scholar] [CrossRef] [PubMed]
- Mammone, T.; Chidlow, G.; Casson, R.J.; Wood, J.P. Expression and activation of mitogen-activated protein kinases in the optic nerve head in a rat model of ocular hypertension. Mol. Cell. Neurosci. 2018, 88, 270–291. [Google Scholar] [CrossRef] [PubMed]
- Dapper, J.D.; Crish, S.D.; Pang, I.-H.; Calkins, D.J. Proximal inhibition of p38 MAPK stress signaling prevents distal axonopathy. Neurobiol. Dis. 2013, 59, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y.; Pang, I.-H.; Shen, J.; Tang, X.; Li, Y.; Liu, C.; Li, B. Protective effect of a JNK inhibitor against retinal ganglion cell loss induced by acute moderate ocular hypertension. Mol. Vis. 2011, 17, 864–875. [Google Scholar] [PubMed]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Libby, R.T. JUN is important for ocular hypertension-induced retinal ganglion cell degeneration. Cell Death Dis. 2017, 8, e2945. [Google Scholar] [CrossRef]
- Liton, P.B.; Li, G.; Luna, C.; Gonzalez, P.; Epstein, D.L. Cross-talk between TGF-β1 and IL-6 in human trabecular meshwork cells. Mol. Vis. 2009, 15, 326–334. [Google Scholar]
- Inoue-Mochita, M.; Inoue, T.; Fujimoto, T.; Kameda, T.; Awai-Kasaoka, N.; Ohtsu, N.; Kimoto, K.; Tanihara, H. p38 MAP Kinase Inhibitor Suppresses Transforming Growth Factor-β2–Induced Type 1 Collagen Production in Trabecular Meshwork Cells. PLoS ONE 2015, 10, e0120774. [Google Scholar] [CrossRef] [PubMed]
- Haddadin, R.I.; Oh, D.-J.; Kang, M.H.; Filippopoulos, T.; Gupta, M.; Hart, L.; Sage, E.H.; Rhee, D.J. SPARC-null Mice Exhibit Lower Intraocular Pressures. Investig. Opthalmol. Vis. Sci. 2009, 50, 3771–3777. [Google Scholar] [CrossRef]
- Oh, D.-J.; Kang, M.H.; Ooi, Y.H.; Choi, K.R.; Sage, E.H.; Rhee, D.J. Overexpression of SPARC in Human Trabecular Meshwork Increases Intraocular Pressure and Alters Extracellular Matrix. Investig. Opthalmol. Vis. Sci. 2013, 54, 3309–3319. [Google Scholar] [CrossRef]
- Kang, M.H.; Oh, D.-J.; Kang, J.-H.; Rhee, D.J. Regulation of SPARC by Transforming Growth Factor β2 in Human Trabecular Meshwork. Investig. Opthalmol. Vis. Sci. 2013, 54, 2523–2532. [Google Scholar] [CrossRef]
- Han, H.; Wecker, T.; Grehn, F.; Schlunck, G. Elasticity-Dependent Modulation of TGF-β Responses in Human Trabecular Meshwork Cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Wecker, T.; Han, H.; Börner, J.; Grehn, F.; Schlunck, G. Effects of TGF-β2 on cadherins and β-catenin in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6456–6462. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, P.P.; Rao, P.V. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am. J. Physiol. Physiol. 2010, 298, C749–C763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Chen, S.D.; Zhang, X.L.; Jonas, J.B. Retinal Microglia in Glaucoma. J. Glaucoma. 2016, 25, 459–465. [Google Scholar] [CrossRef]
- Yu, H.; Zhong, H.; Li, N.; Chen, K.; Chen, J.; Sun, J.; Xu, L.; Wang, J.; Zhang, M.; Liu, X.; et al. Osteopontin activates retinal microglia causing retinal ganglion cells loss via p38 MAPK signaling pathway in glaucoma. FASEB J. 2021, 35, e21405. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Henty-Ridilla, J.L.; Bernstein, A.M.; Ganapathy, P.S.; Herberg, S. TGFβ2 Regulates Human Trabecular Meshwork Cell Contractility via ERK and ROCK Pathways with Distinct Signaling Crosstalk Dependent on the Culture Substrate. Curr. Eye Res. 2022, 47, 1165–1178. [Google Scholar] [CrossRef]
- Irnaten, M.; Duff, A.; Clark, A.; O’Brien, C. Intra-Cellular Calcium Signaling Pathways (PKC, RAS/RAF/MAPK, PI3K) in Lamina Cribrosa Cells in Glaucoma. J. Clin. Med. 2020, 10, 62. [Google Scholar] [CrossRef]
- Lambert, W.S.; Pasini, S.; Collyer, J.W.; Formichella, C.R.; Ghose, P.; Carlson, B.J.; Calkins, D.J. Of Mice and Monkeys: Neuroprotective Efficacy of the p38 Inhibitor BIRB 796 Depends on Model Duration in Experimental Glaucoma. Sci. Rep. 2020, 10, 8535. [Google Scholar] [CrossRef]
- Harder, J.M.; Williams, P.A.; Soto, I.; Foxworth, N.E.; Fernandes, K.A.; Freeburg, N.F.; Libby, R.T.; John, S.W.M. Jnk2 deficiency increases the rate of glaucomatous neurodegeneration in ocular hypertensive DBA/2J mice. Cell Death Dis. 2018, 9, 705. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kao, W.W. Corneal Epithelial Wound Healing. Prog. Mol. Biol. Transl. Sci. 2015, 134, 61–71. [Google Scholar]
- Yu, F.-S.X.; Yin, J.; Xu, K.; Huang, J. Growth factors and corneal epithelial wound healing. Brain Res. Bull. 2010, 81, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Altan, Z.M.; Fenteany, G. c-Jun N-terminal kinase regulates lamellipodial protrusion and cell sheet migration during epithelial wound closure by a gene expression-independent mechanism. Biochem. Biophys. Res. Commun. 2004, 322, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Imayasu, M.; Shimada, S. Phosphorylation of MAP kinase in corneal epithelial cells during wound healing. Curr. Eye Res. 2003, 27, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.D.; He, J.; Bazan, H.E. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: Evidence of cross-talk activation between MAP kinase cascades. J. Biol. Chem. 2003, 278, 21989–21997. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, H.; Tachado, S.; Capo’-Aponte, J.E.; Bildin, V.N.; Koziel, H.; Reinach, P.S. Phosphatase-Mediated Crosstalk Control of ERK and p38 MAPK Signaling in Corneal Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2006, 47, 5267–5275. [Google Scholar] [CrossRef]
- Terai, K.; Call, M.K.; Liu, H.; Saika, S.; Liu, C.Y.; Hayashi, Y.; Chikama, T.; Zhang, J.; Terai, N.; Kao, C.W.; et al. Crosstalk between TGF-beta and MAPK signaling during corneal wound healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8208–8215. [Google Scholar] [CrossRef]
- Saika, S. TGF-beta signal transduction in corneal wound healing as a therapeutic target. Cornea 2004, 23 (Suppl. S8), S25–S30. [Google Scholar] [CrossRef]
- Maeng, Y.-S.; Lee, G.-H.; Lee, B.; Choi, S.-I.; Kim, T.-I.; Kim, E.K. Role of TGFBIp in Wound Healing and Mucin Expression in Corneal Epithelial Cells. Yonsei Med. J. 2017, 58, 423–431. [Google Scholar] [CrossRef]
- Zhong, J.; Hu, N.; Xiong, X.; Lei, Q.; Li, L. A novel promising therapy for skin aging: Dermal multipotent stem cells against photoaged skin by activation of TGF-β/Smad and p38 MAPK signaling pathway. Med. Hypotheses 2011, 76, 343–346. [Google Scholar] [CrossRef]
- Saika, S.; Okada, Y.; Miyamoto, T.; Yamanaka, O.; Ohnishi, Y.; Ooshima, A.; Liu, C.-Y.; Weng, D.; Kao, W.W.-Y. Role of p38 MAP Kinase in Regulation of Cell Migration and Proliferation in Healing Corneal Epithelium. Investig. Opthalmol. Vis. Sci. 2004, 45, 100–109. [Google Scholar] [CrossRef]
- Huh, M.-I.L.; Chang, Y.; Jung, J.-C. Temporal and spatial distribution of TGF-beta isoforms and signaling intermediates in corneal regenerative wound repair. Histol. Histopathol. 2009, 24, 1405–1416. [Google Scholar] [PubMed]
- Nagai, N.; Fukuoka, Y.; Ishii, M.; Otake, H.; Yamamoto, T.; Taga, A.; Okamoto, N.; Shimomura, Y. Instillation of Sericin Enhances Corneal Wound Healing through the ERK Pathway in Rat Debrided Corneal Epithelium. Int. J. Mol. Sci. 2018, 19, 1123. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.-S.; Yoo, Y.-S.; Kwon, J.-Y.; Joo, J.-S.; Lim, S.-A.; Whang, W.-J.; Mok, J.-W.; Choi, J.-S.; Joo, C.-K. Diquafosol promotes corneal epithelial healing via intracellular calcium-mediated ERK activation. Exp. Eye Res. 2016, 143, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Lu, Q.; Teng, Y.; Li, K.; Li, N. Chitosan Promoted the Corneal Epithelial Wound Healing via Activation of ERK Pathway. Curr. Eye Res. 2016, 42, 21–27. [Google Scholar] [CrossRef]
- Mediero, A.; Guzmán-Aranguez, A.; Crooke, A.; Peral, A.; Pintor, J. Corneal re-epithelialization stimulated by diadenosine polyphosphates recruits RhoA/ROCK and ERK1/2 pathways. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4982–4992. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Yi, X.J.; Yu, F.S. Activation of ERK1/2 MAP kinase pathway induces tight junction disruption in human corneal epithelial cells. Exp. Eye. Res. 2004, 78, 125–136. [Google Scholar] [CrossRef]
- Joko, T.; Shiraishi, A.; Akune, Y.; Tokumaru, S.; Kobayashi, T.; Miyata, K.; Ohashi, Y. Involvement of P38MAPK in human corneal endothelial cell migration induced by TGF-β2. Exp. Eye Res. 2012, 108, 23–32. [Google Scholar] [CrossRef]
- Nakahara, M.; Okumura, N.; Nakano, S.; Koizumi, N. Effect of a p38 Mitogen-Activated Protein Kinase Inhibitor on Corneal Endothelial Cell Proliferation. Investig. Opthalmol. Vis. Sci. 2018, 59, 4218–4227. [Google Scholar] [CrossRef]
- Hongo, A.; Okumura, N.; Nakahara, M.; Kay, E.P.; Koizumi, N. The Effect of a p38 Mitogen-Activated Protein Kinase Inhibitor on Cellular Senescence of Cultivated Human Corneal Endothelial Cells. Investig. Opthalmol. Vis. Sci. 2017, 58, 3325–3334. [Google Scholar] [CrossRef]
- Mao, Y.; Ou, S.; Zhu, C.; Lin, S.; Liu, X.; Liang, M.; Yu, J.; Wu, Y.; He, H.; Zong, R.; et al. Downregulation of p38 MAPK Signaling Pathway Ameliorates Tissue-Engineered Corneal Epithelium. Tissue Eng. Part A 2022, 28, 977–989. [Google Scholar] [CrossRef]
- Okada, Y.; Saika, S.; Shirai, K.; Yamanaka, O.; Kitano, A.; Wang, Z.; Yang, H.; Reinach, P. JNK MAPK Signaling Contributes in vivo to Injury-Induced Corneal Epithelial Migration. Ophthalmic Res. 2009, 42, 185–192. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, L.-Y.; Li, C.-Y.; Wu, J.-Y.; Zhang, Y.-T.; Pang, K.-P.; Wei, Y.; Du, L.-Q.; Liu, M.; Wu, X.-Y. SPARC promotes self-renewal of limbal epithelial stem cells and ocular surface restoration through JNK and p38-MAPK signaling pathways. Stem Cells 2019, 38, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chang, Y.; Yang, Y.; Zhang, Y.; Yu, F.-S.X.; Wu, X. Activation of JNK Signaling Mediates Connective Tissue Growth Factor Expression and Scar Formation in Corneal Wound Healing. PLoS ONE 2012, 7, e32128. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Zhang, Y.; Zhang, L.; Yeh, L.-K.; Wang, Y.-C.; Saika, S.; Liu, C.-Y. Shp2-mediated MAPK pathway regulates ΔNp63 in epithelium to promote corneal innervation and homeostasis. Lab. Investig. 2019, 100, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-C.; Huh, M.-I.; Fini, M.E. Constitutive collagenase-1 synthesis through MAPK pathways is mediated, in part, by endogenous IL-1α during fibrotic repair in corneal stroma. J. Cell. Biochem. 2007, 102, 453–462. [Google Scholar] [CrossRef]
- Chao, S.-C.; Nien, C.-W.; Iacob, C.; Hu, D.-N.; Huang, S.-C.; Lin, H.-Y. Effects of Lutein on Hyperosmoticity-Induced Upregulation of IL-6 in Cultured Corneal Epithelial Cells and Its Relevant Signal Pathways. J. Ophthalmol. 2016, 2016, 8341439. [Google Scholar] [CrossRef]
- Luo, L.; Li, D.-Q.; Doshi, A.; Farley, W.; Corrales, R.M.; Pflugfelder, S.C. Experimental Dry Eye Stimulates Production of Inflammatory Cytokines and MMP-9 and Activates MAPK Signaling Pathways on the Ocular Surface. Investig. Opthalmol. Vis. Sci. 2004, 45, 4293–4301. [Google Scholar] [CrossRef]
- De Paiva, C.S.; Corrales, R.M.; Villarreal, A.L.; Farley, W.J.; Li, D.-Q.; Stern, M.E.; Pflugfelder, S.C. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp. Eye Res. 2006, 83, 526–535. [Google Scholar] [CrossRef]
- Wu, Y.; Bu, J.; Yang, Y.; Lin, X.; Cai, X.; Huang, C.; Zheng, X.; Ouyang, W.; Li, W.; Zhang, X.; et al. Therapeutic Effect of MK2 Inhibitor on Experimental Murine Dry Eye. Investig. Opthalmol. Vis. Sci. 2017, 58, 4898–4907. [Google Scholar] [CrossRef]
- Jiang, D.; Liu, X.; Hu, J. Topical administration of Esculetin as a potential therapy for experimental dry eye syndrome. Eye 2017, 31, 1724–1732. [Google Scholar] [CrossRef]
- Ling, J.; Chan, C.L.; Ho, C.Y.; Gao, X.; Tsang, S.M.; Leung, P.C.; Hu, J.M.; Wong, C.K. The Extracts of Dendrobium Alleviate Dry Eye Disease in Rat Model by Regulating Aquaporin Expression and MAPKs/NF-κB Signalling. Int. J. Mol. Sci. 2022, 23, 11195. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, T.; Shivakumar, S.; Shetty, R.; D’Souza, S.; Nelson, E.J.R.; Sethu, S.; Jeyabalan, N.; Ghosh, A. Trehalose augments autophagy to mitigate stress induced inflammation in human corneal cells. Ocul. Surf. 2019, 17, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Khaled, M.L.; McKay, T.B.; Liu, Y.; Karamichos, D. Transcriptional profiling of corneal stromal cells derived from patients with keratoconus. Sci. Rep. 2019, 9, 12567. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.-D.; Gao, H.; Xu, W.-H.; Shan, C.; Liu, Y.; Zhou, Z.-X.; Wang, K.; Li, P.-F. Systematically Displaying the Pathogenesis of Keratoconus via Multi-Level Related Gene Enrichment-Based Review. Front. Med. 2022, 8, 770138. [Google Scholar] [CrossRef] [PubMed]
- Landsend, E.C.; Lagali, N.; Utheim, T.P. Congenital aniridia—A comprehensive review of clinical features and therapeutic approaches. Surv. Ophthalmol. 2021, 66, 1031–1050. [Google Scholar] [CrossRef] [PubMed]
- Alkatan, H.M.; Al Dhaheri, H.; Al Harby, M. Terminology of Peters’ anomaly variants: Summary of histopathological findings in 6 corneas and detailed clinicopathological correlation in 2 cases. Saudi J. Ophthalmol. 2018, 33, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Latta, L.; Figueiredo, F.; Ashery-Padan, R.; Collinson, J.; Daniels, J.; Ferrari, S.; Szentmáry, N.; Solá, S.; Shalom-Feuerstein, R.; Lako, M.; et al. Pathophysiology of aniridia-associated keratopathy: Developmental aspects and unanswered questions. Ocul. Surf. 2021, 22, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Leiper, L.J.; Walczysko, P.; Kucerova, R.; Ou, J.; Shanley, L.J.; Lawson, D.; Forrester, J.V.; McCaig, C.D.; Zhao, M.; Collinson, J.M. The roles of calcium signaling and ERK1/2 phosphorylation in a Pax6+/− mouse model of epithelial wound-healing delay. BMC Biol. 2006, 4, 27. [Google Scholar] [CrossRef]
- Latta, L.; Ludwig, N.; Krammes, L.; Stachon, T.; Fries, F.; Mukwaya, A.; Szentmáry, N.; Seitz, B.; Wowra, B.; Kahraman, M.; et al. Abnormal neovascular and proliferative conjunctival phenotype in limbal stem cell deficiency is associated with altered microRNA and gene expression modulated by PAX6 mutational status in congenital aniridia. Ocul. Surf. 2021, 19, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Lowes, C.; Collinson, J.M. Cytoskeletal and cell adhesion defects in wounded and Pax6+/− corneal epithelia. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1415–1423. [Google Scholar] [CrossRef]
- Rabiee, B.; Anwar, K.N.; Shen, X.; Putra, I.; Liu, M.; Jung, R.; Afsharkhamseh, N.; Rosenblatt, M.I.; Fishman, G.A.; Liu, X.; et al. Gene dosage manipulation alleviates manifestations of hereditary PAX6 haploinsufficiency in mice. Sci. Transl. Med. 2020, 12, eaaz4894. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.D.; McHaney, K.M.; Rabiee, B.; Gao, J.; Rodriguez, C.; Miller, D.A.; Liu, M.; Grannonico, M.; Norat, P.; Zhang, H.F.; et al. Long-term retinal protection by MEK inhibition in Pax6 haploinsufficiency mice. Exp. Eye Res. 2022, 218, 109012. [Google Scholar] [CrossRef]
- Roux, L.N.; Petit, I.; Domart, R.; Concordet, J.-P.; Qu, J.; Zhou, H.; Joliot, A.; Ferrigno, O.; Aberdam, D. Modeling of Aniridia-Related Keratopathy by CRISPR/Cas9 Genome Editing of Human Limbal Epithelial Cells and Rescue by Recombinant PAX6 Protein. Stem Cells 2018, 36, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Oved, K.; Zennaro, L.; Dorot, O.; Zerbib, J.; Frank, E.; Roux, L.N.; Bremond-Gignac, D.; Pichinuk, E.; Aberdam, D. Ritanserin, a potent serotonin 2A receptor antagonist, represses MEK/ERK signalling pathway to restore PAX6 production and function in aniridia-like cellular model. Biochem. Biophys. Res. Commun. 2021, 582, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Dorot, O.; Roux, L.N.; Zennaro, L.; Oved, K.; Bremond-Gignac, D.; Pichinuk, E.; Aberdam, D. The antipsychotropic drug Duloxetine rescues PAX6 haploinsufficiency of mutant limbal stem cells through inhibition of the MEK/ERK signaling pathway. Ocul. Surf. 2022, 23, 140–142. [Google Scholar] [CrossRef]
- Li, T.; Lu, L. Epidermal Growth Factor-induced Proliferation Requires Down-regulation of Pax6 in Corneal Epithelial Cells. J. Biol. Chem. 2005, 280, 12988–12995. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.L.; Mist, S.D.; Ross, R.L.; Jones, K.D. Duloxetine for the treatment of fibromyalgia. Expert Rev. Clin. Immunol. 2010, 6, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Daneshpajooh, A.; Anvari, S.O.; Dozchizadeh, S.; Teimorian, M. Evaluation of the Clinical Efficacy and Complications of Duloxetine in Comparison to Solifenacin in the Treatment of Overactive Bladder Disease in Women: A Randomized Clinical Trial. Urol. J. 2021, 18, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Drugs.com. Cymbalta Package Insert—Prescribing Information. 2022. Available online: https://www.drugs.com/pro/cymbalta.html (accessed on 26 December 2022).
- Drugs.com. Duloxetine Side Effects. 2022. Available online: https://www.drugs.com/sfx/duloxetine-side-effects.html (accessed on 26 December 2022).
- Hu, D.-N.; Bi, M.; Zhang, D.Y.; Ye, F.; McCormick, S.A.; Chan, C.-C. Constitutive and LPS-Induced Expression of MCP-1 and IL-8 by Human Uveal Melanocytes In Vitro and Relevant Signal Pathways. Investig. Opthalmol. Vis. Sci. 2014, 55, 5760–5769. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.N.; Chen, M.; Zhang, D.Y.; Ye, F.; McCormick, S.A.; Chan, C.C. Interleukin-1beta increases baseline expression and secretion of interleukin-6 by human uveal melanocytes in vitro via the p38 MAPK/NF-κB pathway. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3767–3774. [Google Scholar] [CrossRef] [PubMed]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis. Life 2021, 12, 33. [Google Scholar] [CrossRef]
- Takeda, M.; Takamiya, A.; Yoshida, A.; Kiyama, H. Extracellular signal-regulated kinase activation predominantly in Müller cells of retina with endotoxin-induced uveitis. Investig. Opthalmol. Vis. Sci. 2002, 43, 907–1011. [Google Scholar]
- Zheng, C.; Lei, C.; Chen, Z.; Zheng, S.; Yang, H.; Qiu, Y.; Lei, B. Topical administration of diminazene aceturate decreases inflammation in endotoxin-induced uveitis. Mol. Vis. 2015, 21, 403–411. [Google Scholar]
- Touchard, E.; Omri, S.; Berdugo, M.; Deloche, C.; Abadie, C.; Jonet, L.; Crisanti, P.; De Kozak, Y.; Behar-Cohen, F.; Naud, M.-C.; et al. A Peptide Inhibitor of c-Jun N-Terminal Kinase for the Treatment of Endotoxin-Induced Uveitis. Investig. Opthalmol. Vis. Sci. 2010, 51, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- El Zaoui, I.; Touchard, E.; Berdugo, M.; Abadie, C.; Kowalczuk, L.; Deloche, C.; Zhao, M.; Naud, M.-C.; Combette, J.-M.; Behar-Cohen, F. Subconjunctival Injection of XG-102, a c-Jun N-Terminal Kinase Inhibitor Peptide, in the Treatment of Endotoxin-Induced Uveitis in Rats. J. Ocul. Pharmacol. Ther. 2015, 31, 17–24. [Google Scholar] [CrossRef]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.-P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.-A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2016, 174, 76–84. [Google Scholar] [CrossRef][Green Version]
- Deloche, C.; Lopez-Lazaro, L.; Mouz, S.; Perino, J.; Abadie, C.; Combette, J.M. XG-102 administered to healthy male volunteers as a single intravenous infusion: A randomized, double-blind, placebo-controlled, dose-escalating study. Pharmacol. Res. Perspect. 2014, 2, e00020. [Google Scholar] [CrossRef]
- Beydoun, T.; Deloche, C.; Perino, J.; Kirwan, B.-A.; Combette, J.-M.; Behar-Cohen, F. Subconjunctival Injection of XG-102, a JNK Inhibitor Peptide, in Patients with Intraocular Inflammation: A Safety and Tolerability Study. J. Ocul. Pharmacol. Ther. 2015, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Sarny, S.; Neumayer, M.; Kofler, J.; El-Shabrawi, Y. Ocular toxicity due to Trametinib and Dabrafenib. BMC Ophthalmol. 2017, 17, 146. [Google Scholar] [CrossRef]
- Rueda-Rueda, T.; Sánchez-Vicente, J.; Moruno-Rodríguez, A.; Molina-Socola, F.; Martínez-Borrego, A.; López-Herrero, F. Uveitis and serous retinal detachment secondary to systemic dabrafenib and trametinib. Arch. Soc. Española Oftalmol. (Engl. Ed.) 2018, 93, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Draganova, D.D.; Kerger, J.; Caspers, L.; Willermain, F. Severe bilateral panuveitis during melanoma treatment by Dabrafenib and Trametinib. J. Ophthalmic Inflamm. Infect. 2015, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Rali, A.; Huang, Y.B.; Yeh, S. Cancer Immunotherapy and Uveitis: Balancing Anti-Tumor Immunity and Ocular Autoimmunity. Int. Ophthalmol. Clin. 2022, 62, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, D.; Ogata, M.; Reneker, L.W. MAPK1 is required for establishing the pattern of cell proliferation and for cell survival during lens development. Development 2013, 140, 1573–1582. [Google Scholar] [CrossRef]
- Peng, J.; Zheng, T.-T.; Liang, Y.; Duan, L.-F.; Zhang, Y.-D.; Wang, L.-J.; He, G.-M.; Xiao, H.-T. p-Coumaric Acid Protects Human Lens Epithelial Cells against Oxidative Stress-Induced Apoptosis by MAPK Signaling. Oxid. Med. Cell. Longev. 2018, 2018, 8549052. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yao, K.; Zhang, L.; Yang, Y.; Yao, H. Honokiol inhibits H2O2-induced apoptosis in human lens epithelial cells via inhibition of the mitogen-activated protein kinase and Akt pathways. Eur. J. Pharmacol. 2011, 650, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-N.; Ge, M.-X.; Yuan, Z.-F. MicroRNA-182-5p protects human lens epithelial cells against oxidative stress-induced apoptosis by inhibiting NOX4 and p38 MAPK signalling. BMC Ophthalmol. 2020, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Shao, J.; Xie, D.; Zhang, F. Decorin inhibits glucose-induced lens epithelial cell apoptosis via suppressing p22phox-p38 MAPK signaling pathway. PLoS ONE 2020, 15, e0224251. [Google Scholar] [CrossRef]
- Bai, J.; Zheng, Y.; Dong, L.; Cai, X.; Wang, G.; Liu, P. Inhibition of p38 mitogen-activated protein kinase phosphorylation decreases H2O2-induced apoptosis in human lens epithelial cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhang, L.; Zhang, Y.; Ye, P.; Zhu, N. The flavonoid, fisetin, inhibits UV radiation-induced oxidative stress and the activation of NF-κB and MAPK signaling in human lens epithelial cells. Mol. Vis. 2008, 14, 1865–1871. [Google Scholar]
- Jia, Z.; Song, Z.; Zhao, Y.; Wang, X.; Liu, P. Grape seed proanthocyanidin extract protects human lens epithelial cells from oxidative stress via reducing NF-κB and MAPK protein expression. Mol. Vis. 2011, 17, 210–217. [Google Scholar]
- Li, X.; Meng, F.; Li, H.; Hua, X.; Wu, L.; Yuan, X. L-carnitine alleviates oxidative stress-related damage via MAPK signaling in human lens epithelial cells exposed to H2O2. Int. J. Mol. Med. 2019, 44, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, W. Sanguinarine induces apoptosis of human lens epithelial cells by increasing reactive oxygen species via the MAPK signaling pathway. Mol. Med. Rep. 2019, 19, 4449–4456. [Google Scholar] [CrossRef]
- Xiao, X.; Zheng, Y.; Mo, Y.; Wang, W.; Li, X.; Wang, J. Astragaloside IV alleviates oxidative stress-related damage via inhibiting NLRP3 inflammasome in a MAPK signaling dependent pathway in human lens epithelial cells. Drug Dev. Res. 2022, 83, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Dong, J. Diosmetin attenuates oxidative stress-induced damage to lens epithelial cells via the mitogen-activated protein kinase (MAPK) pathway. Bioengineered 2022, 13, 11072–11081. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Fang, C. Knockdown of Tripartite motif-containing 22 (TRIM22) relieved the apoptosis of lens epithelial cells by suppressing the expression of TNF receptor-associated factor 6 (TRAF6). Bioengineered 2021, 12, 7213–7222. [Google Scholar] [CrossRef]
- Jia, Y.; Qin, Q.; Fang, C.P.; Shen, W.; Sun, T.T.; Huang, Y.L.; Li, W.J.; Deng, A.M. UVB induces apoptosis via downregulation of CALML3-dependent JNK1/2 and ERK1/2 pathways in cataract. Int. J. Mol. Med. 2018, 41, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ma, Y.; Xu, Y. Taxifolin Shows Anticataractogenesis and Attenuates Diabetic Retinopathy in STZ-Diabetic Rats via Suppression of Aldose Reductase, Oxidative Stress, and MAPK Signaling Pathway. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 599–608. [Google Scholar] [CrossRef]
- Zhang, P.; Xing, K.; Randazzo, J.; Blessing, K.; Lou, M.F.; Kador, P.F. Osmotic stress, not aldose reductase activity, directly induces growth factors and MAPK signaling changes during sugar cataract formation. Exp. Eye Res. 2012, 101, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Hashida, N.; Ping, X.; Nishida, K. MAPK activation in mature cataract associated with Noonan syndrome. BMC Ophthalmol. 2013, 13, 70. [Google Scholar] [CrossRef]
- Méndez-Martínez, S.; Calvo, P.; Ruiz-Moreno, O.; Barón, N.P.; Bueno, J.L.; Ruiz, M.D.R.G.; Pablo, L. Ocular Adverse Events Associated with Mek Inhibitors. Retina 2019, 39, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Fauviaux, E.; Promelle, V.; Boucenna, V.; Jany, B.; Errera, M.H.; Delbarre, M.; Boucenna, W. Ocular toxicity of targeted therapies with MEK inhibitors and BRAF inhibitors in the treatment of metastatic cutaneous melanoma. J. Fr. Ophtalmol. 2022, 45, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Niro, A.; Strippoli, S.; Alessio, G.; Sborgia, L.; Recchimurzo, N.; Guida, M. Ocular Toxicity in Metastatic Melanoma Patients Treated with Mitogen-Activated Protein Kinase Kinase Inhibitors: A Case Series. Am. J. Ophthalmol. 2015, 160, 959–967.e1. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Habib, L.A.; Abramson, D.H.; Yannuzzi, L.A.; Heinemann, M.; Gounder, M.M.; Grisham, R.N.; Postow, M.A.; Shoushtari, A.N.; Chi, P.; et al. Clinical and Morphologic Characteristics of MEK Inhibitor-Associated Retinopathy: Differences from Central Serous Chorioretinopathy. Ophthalmology 2017, 124, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Nti, A.A.; Serrano, L.W.; Sandhu, H.S.; Uyhazi, K.E.; Edelstein, I.D.; Zhou, E.J.; Bowman, S.; Song, D.; Gangadhar, T.C.; Schuchter, L.M.; et al. Frequent Subclinical Macular Changes in Combined Braf/Mek Inhibition with High-Dose Hydroxychloroquine as Treatment for Advanced Metastatic Braf Mutant Melanoma: Preliminary Results from a Phase I/Ii Clinical Treatment Trial. Retina 2019, 39, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Urner-Bloch, U.; Urner, M.; Jaberg-Bentele, N.; Frauchiger, A.L.; Dummer, R.; Goldinger, S.M. MEK inhibitor-associated retinopathy (MEKAR) in metastatic melanoma: Long-term ophthalmic effects. Eur. J. Cancer 2016, 65, 130–138. [Google Scholar] [CrossRef]
- van Dijk, E.H.; van Herpen, C.M.; Marinkovic, M.; Haanen, J.B.; Amundson, D.; Luyten, G.P.; Jager, M.J.; Kapiteijn, E.H.; Keunen, J.E.; Adamus, G.; et al. Serous Retinopathy Associated with Mitogen-Activated Protein Kinase Kinase Inhibition (Binimetinib) for Metastatic Cutaneous and Uveal Melanoma. Ophthalmology 2015, 122, 1907–1916. [Google Scholar] [CrossRef]
- Weber, M.L.; Liang, M.C.; Flaherty, K.T.; Heier, J.S. Subretinal Fluid Associated with MEK Inhibitor Use in the Treatment of Systemic Cancer. JAMA Ophthalmol. 2016, 134, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Santiago, C. New features in MEK retinopathy. BMC Ophthalmol. 2018, 18 (Suppl. S1), 221. [Google Scholar] [CrossRef]
- Van Dijk, E.H.; Kruit, W.H.; Jager, M.J.; Luyten, G.P.; Vingerling, J.R.; Boon, C.J. Pimasertib-associated ophthalmological adverse events. Acta Ophthalmol. 2018, 96, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Barteselli, G.; Goodman, G.R.; Patel, Y.; Caro, I.; Xue, C.; McCallum, S. Characterization of Serous Retinopathy Associated with Cobimetinib: Integrated Safety Analysis of Four Studies. Drug Saf. 2022, 45, 1491–1499. [Google Scholar] [CrossRef]
- Francis, J.H.; Canestraro, J.; Haggag-Lindgren, D.; Harding, J.J.; Diamond, E.L.; Drilon, A.; Li, B.T.; Iyer, G.; Schram, A.M.; Abramson, D.H. Clinical and Morphologic Characteristics of Extracellular Signal-Regulated Kinase Inhibitor-Associated Retinopathy. Ophthalmol. Retin. 2021, 5, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Mettler, C.; Monnet, D.; Kramkimel, N.; Tréluyer, J.M.; Mouthon, L.; Brézin, A.; Dupin, N.; Valnet-Rabier, M.B.; Chouchana, L.; Terrier, B. Ocular Safety Profile of BRAF and MEK Inhibitors: Data from the World Health Organization Pharmacovigilance Database. Ophthalmology 2021, 128, 1748–1755. [Google Scholar] [CrossRef] [PubMed]
- Brambati, M.; Giuffrè, C.; Marchese, A.; Bandello, F.; Modorati, G.M.; Miserocchi, E. A case of Vogt-Koyanagi-Harada-like uveitis secondary to dabrafenib/trametinib therapy for advanced melanoma. Eur. J. Ophthalmol. 2022, 32, NP109–NP113. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, N.; Velazquez-Martin, J.P.; Bedard, P.L. Ocular toxicities of MEK inhibitors and other targeted therapies. Ann. Oncol. 2016, 27, 998–1005. [Google Scholar] [CrossRef]
- Delord, J.P.; Italiano, A.; Awada, A.; Aftimos, P.; Houédé, N.; Lebbé, C.; Pages, C.; Lesimple, T.; Dinulescu, M.; Schellens, J.H.M.; et al. Selective Oral MEK1/2 Inhibitor Pimasertib: A Phase I Trial in Patients with Advanced Solid Tumors. Target Oncol. 2021, 16, 37–46. [Google Scholar] [CrossRef]
- Drugs.com. Selumetinib Side Effects. 2022. Available online: https://www.drugs.com/sfx/selumetinib-side-effects.html (accessed on 9 January 2023).
- Murali, R.; Menzies, A.M.; Long, G. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2012, 6, 391–405. [Google Scholar] [CrossRef]
- Drugs.com. Dabrafenib Side Effects. 2022. Available online: https://www.drugs.com/sfx/dabrafenib-side-effects.html (accessed on 9 January 2023).



| Gene Name | Protein Name | Alternative Protein Names | Pathway Involved | MAPK Level | Other Gene/Protein Names |
|---|---|---|---|---|---|
| MAPK1 | ERK2 | p42-MAPK | MEK/ERK | MAPK | MAPK2, p38, p40, p41, ERT1, NS13 |
| MAPK3 | ERK1 | p44-MAPK | MEK/ERK | MAPK | ERT2, PRKM3 |
| MAPK4 | ERK4 | p63-MAPK | atypical MAPK | MAPK | PRKM4 |
| MAPK6 | ERK3 | p97-MAPK | atypical MAPK | MAPK | PRKM6, HsT17250 |
| MAPK7 | ERK5 | ERK5 | MAPK | PRKM7, BMK1 | |
| MAPK8 | JNK1 | SAPK1 | JNK | MAPK | PRKM8 |
| MAPK9 | JNK2 | p54aSAPK | JNK | MAPK | PRKM9 |
| MAPK10 | JNK3 | p54bSAPK | JNK | MAPK | PRKM10, SAPK1b, p493F12 |
| MAPK11 | p38 beta | SAPK2, SAPK2B | p38 | MAPK | PRKM11 |
| MAPK12 | p38 gamma | ERK6, SAPK-3 | p38 | MAPK | PRKM12 |
| MAPK13 | p38 delta | SAPK4 | p38 | MAPK | PRKM13 |
| MAPK14 | p38 alpha | SAPK2A, Mxi2 | p38 | MAPK | PRKM14, PRKM15, CSBP, EXIP |
| MAPK15 | ERK7/8 | atypical MAPK | MAPK | ||
| MAP2K1 | MEK1 | MKK1, MAPKK1 | MEK/ERK | MAP2K | CFC3 |
| MAP2K2 | MEK2 | MKK2, MAPKK2 | MEK/ERK | MAP2K | CFC4 |
| MAP2K3 | MEK3 | MKK3, MAPKK3 | p38 | MAP2K | SAPKK2 |
| MAP2K4 | MEK4 | MKK4, MAPKK4 | JNK | MAP2K | SAPKK1, JNKK1, JNKK |
| MAP2K5 | MEK5 | MAPKK5 | ERK5 | MAP2K | |
| MAP2K6 | MEK6 | MKK6, MAPKK6 | p38 | MAP2K | SAPKK3 |
| MAP2K7 | MEK7 | MKK7, MAPKK7 | JNK | MAP2K | SAPKK4, JNKK2 |
| RAF1 | c-Raf | Raf-1 | MEK/ERK | MAP3K | |
| BRAF | B-Raf | BRAF-1, RAFB1 | MEK/ERK | MAP3K | NS7 |
| MAP3K1 | MEKK1 | JNK | MAP3K | ||
| MAP3K2 | MEKK2 | MEKK2B | ERK5 | MAP3K | |
| MAP3K3 | MEKK3 | MAPKKK3 | ERK5 | MAP3K | |
| MAP3K4 | MEKK4 | MAPKKK4 | MAP3K | MTK1, PRO0412 | |
| MAP3K5 | ASK1 | MEKK5, MAPKKK5 | JNK and p38 | MAP3K | |
| MAP3K6 | ASK2 | MEKK6, MAPKKK6 | MAP3K | ||
| MAP3K7 | TAK1 | MEKK7, TGF1a | JNK and p38 | MAP3K | CSCF, FMD2 |
| MAP3K8 | MEKK8 | Tpl-2, c-COT | MAP3K | COT, EST, ESTF, AURA2 | |
| MAP3K9 | MLK1 | MEKK9 | MAP3K | PRKE1 | |
| MAP3K10 | MLK2 | MEKK10 | MAP3K | MST | |
| MAP3K11 | MLK3 | MEKK11 | JNK and p38 | MAP3K | PTK1, SPRK |
| MAP3K12 | ZPK | MEKK12 | MAP3K | DLK, MUK, HP09298 | |
| MAP3K13 | LZK | MEKK13 | JNK | MAP3K | MLK |
| MAP3K14 | MAP3K | FTDCR1B, HS, HSNIK, NIK | |||
| MAP3K15 | ASK3 | MAP3K | bA723P2.3 | ||
| TAOK1 | PSK2 | MAP3K16, TAO1 | JNK | MAP3K | DDIB, KFC-B, MARKK, hKFC-B |
| TAOK2 | PSK | MAP3K17, TAO2 | MAP3K | Tao2beta, PSK1-BETA | |
| TAOK3 | MAP3K18 | p38 | MAP3K | DPK, JIK, hKFC-A | |
| MAP3K19 | MAP3K | RCK, YSK4 | |||
| MAP3K20 | MLK7 | mlklak, pk | MAP3K | AZK, MLT, MRK, ZAK, SFMMP | |
| MAP3K21 | MLK4 | dJ862P8.3 | MAP3K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustardas, P.; Aberdam, D.; Lagali, N. MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges. Cells 2023, 12, 617. https://doi.org/10.3390/cells12040617
Moustardas P, Aberdam D, Lagali N. MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges. Cells. 2023; 12(4):617. https://doi.org/10.3390/cells12040617
Chicago/Turabian StyleMoustardas, Petros, Daniel Aberdam, and Neil Lagali. 2023. "MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges" Cells 12, no. 4: 617. https://doi.org/10.3390/cells12040617
APA StyleMoustardas, P., Aberdam, D., & Lagali, N. (2023). MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges. Cells, 12(4), 617. https://doi.org/10.3390/cells12040617