


Role of C-Terminal Phosphorylation of Lamin A in DNA Damage and Cellular Senescence

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Mutants and MEF Culture
2.2. Plasmid Construction
- LMNA-S625A-F: 5′-CCACACTGCGGTAGGCGCGAGTGACCGTGA-3′
- LMNA-S625A-R: 5′-TCACGGTCACTCGCGCCTACCGCAGTGTGG-3′
- LMNA-S628A-F: 5′-ACTGCCCCCCACAGCGCGGTAGCTGCGA-3
- LMNA-S628A-R: 5′-TCGCAGCTACCGCGCTGTGGGGGGCAGT-3′
- LMNA-S632A-F: 5′-GCTGCCACCCCCAGCGCCCCCCACACTG-3
- LMNA-S632A-R: 5′-CAGTGTGGGGGGCGCTGGGGGTGGCAGC-3′
- LMNA-S636A-F: 5′-GCAGTGGGGGTGGCGCCTTCGGGGACAATC-3′
- LMNA-S636A-R: 5′-GATTGTCCCCGAAGGCGCCACCCCCACTGC-3′
- LMNA-S652A-F: 5′-CTGGGTTCGGGGGGCGGAGTTGCCCAGG-3′
- LMNA-S652A-R: 5′-CCTGGGCAACTCCGCCCCCCGAACCCAG-3′
2.3. Antibodies
2.4. Western Blotting
2.5. Immunoprecipitation
2.6. Protein Purification and Pull-Down Assays
2.7. In Vitro Kinase Assay
2.8. Senescence-Associated β-Gal Assay
2.9. Statistical Analyses
3. Results
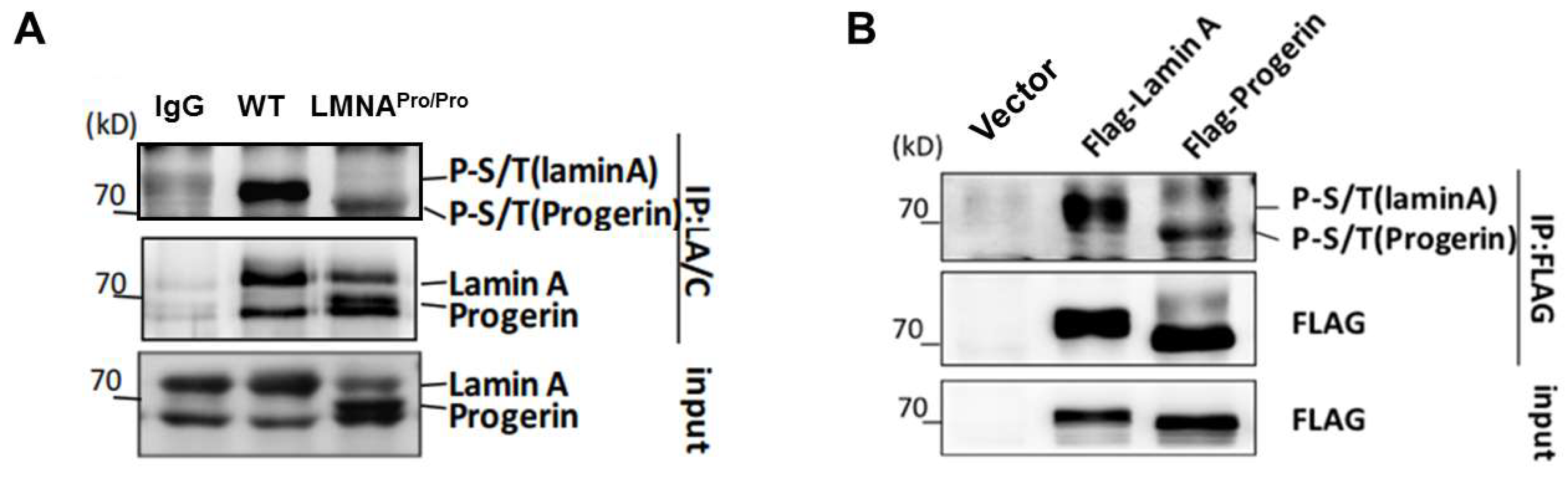
3.1. Lower Level of Phosphorylation in Progerin Compared with Lamin A
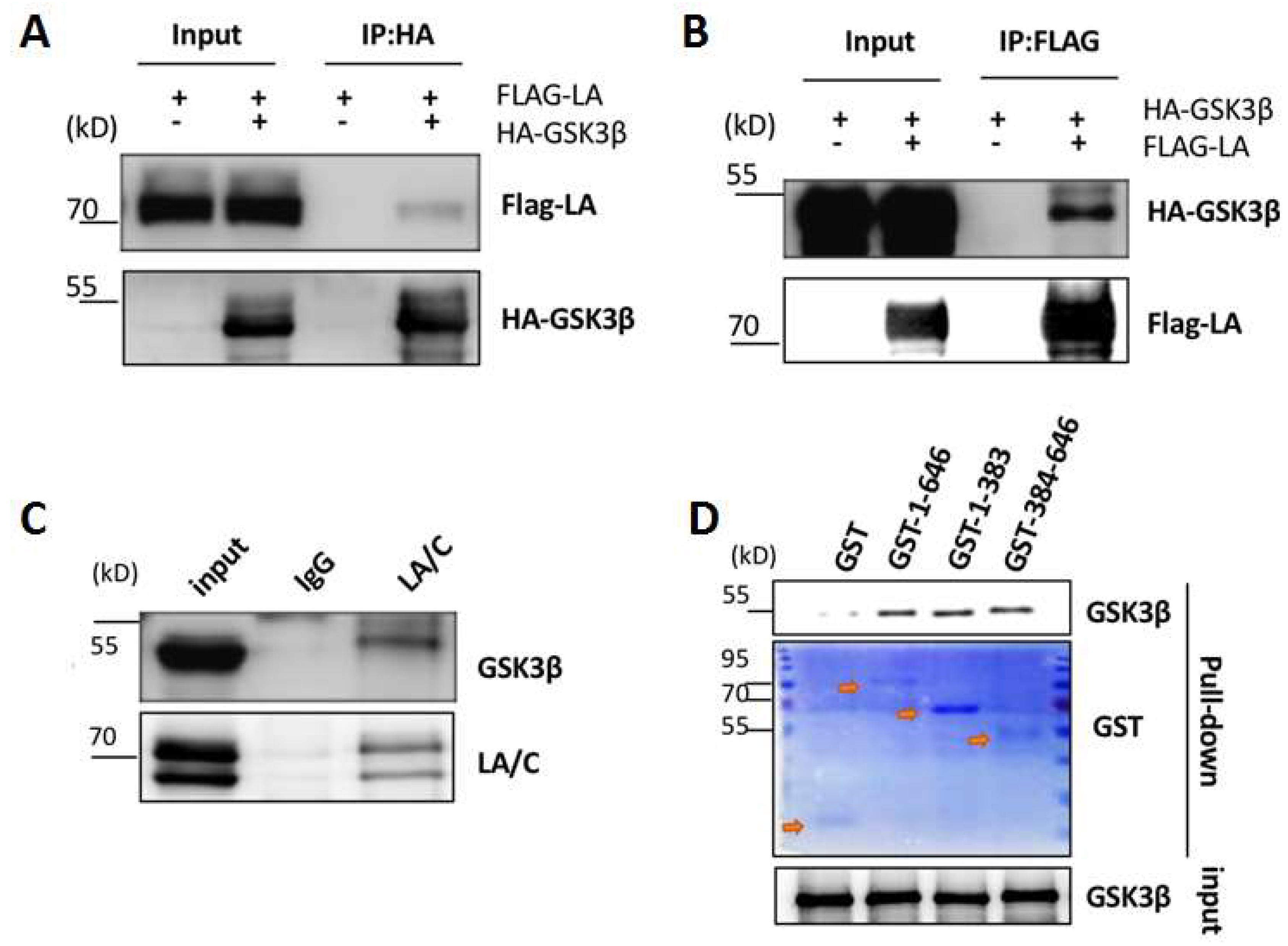
3.2. Interaction between Lamin A and GSK3β
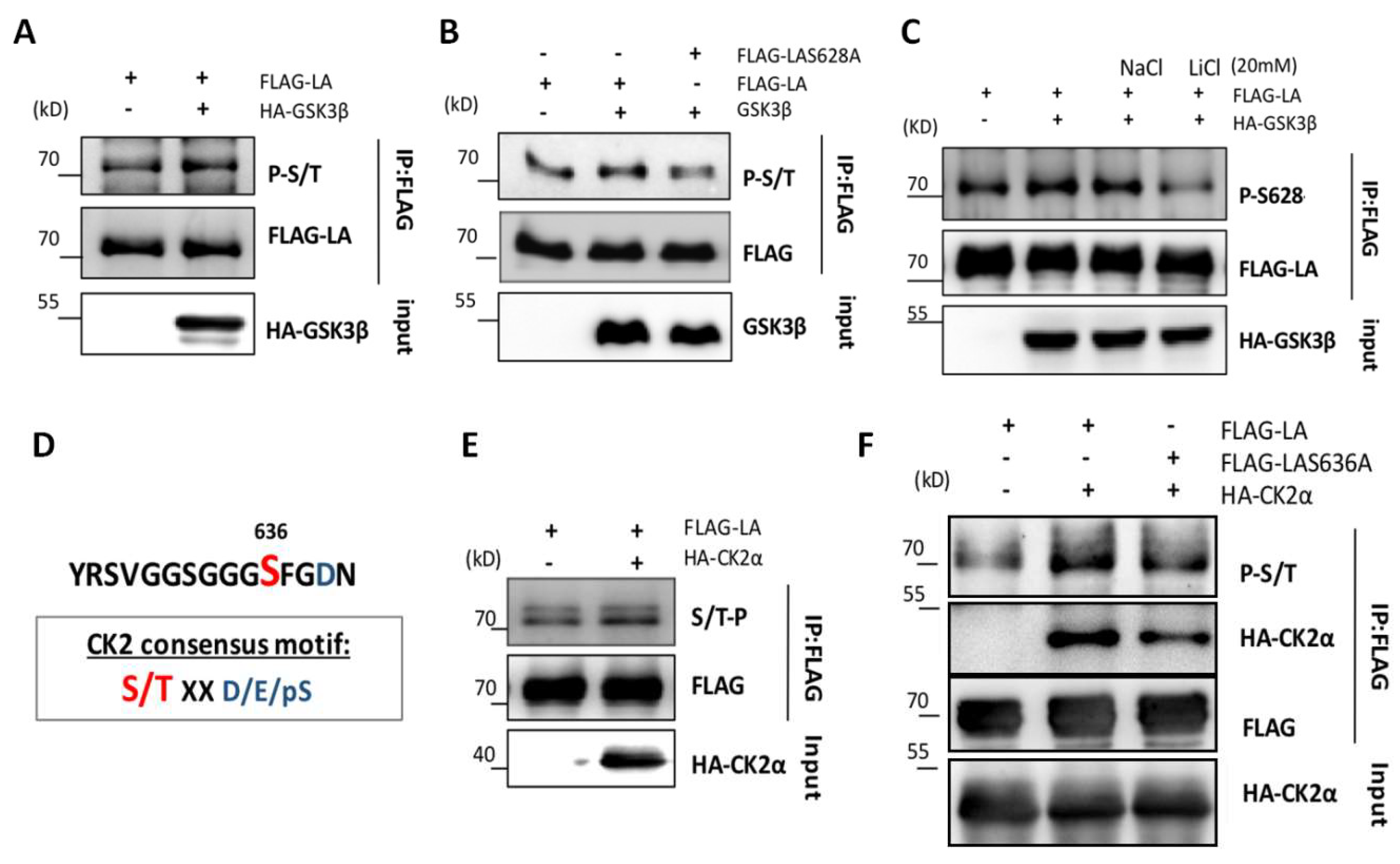
3.3. Role of GSK3β in the Phosphorylation of Lamin A at Ser628
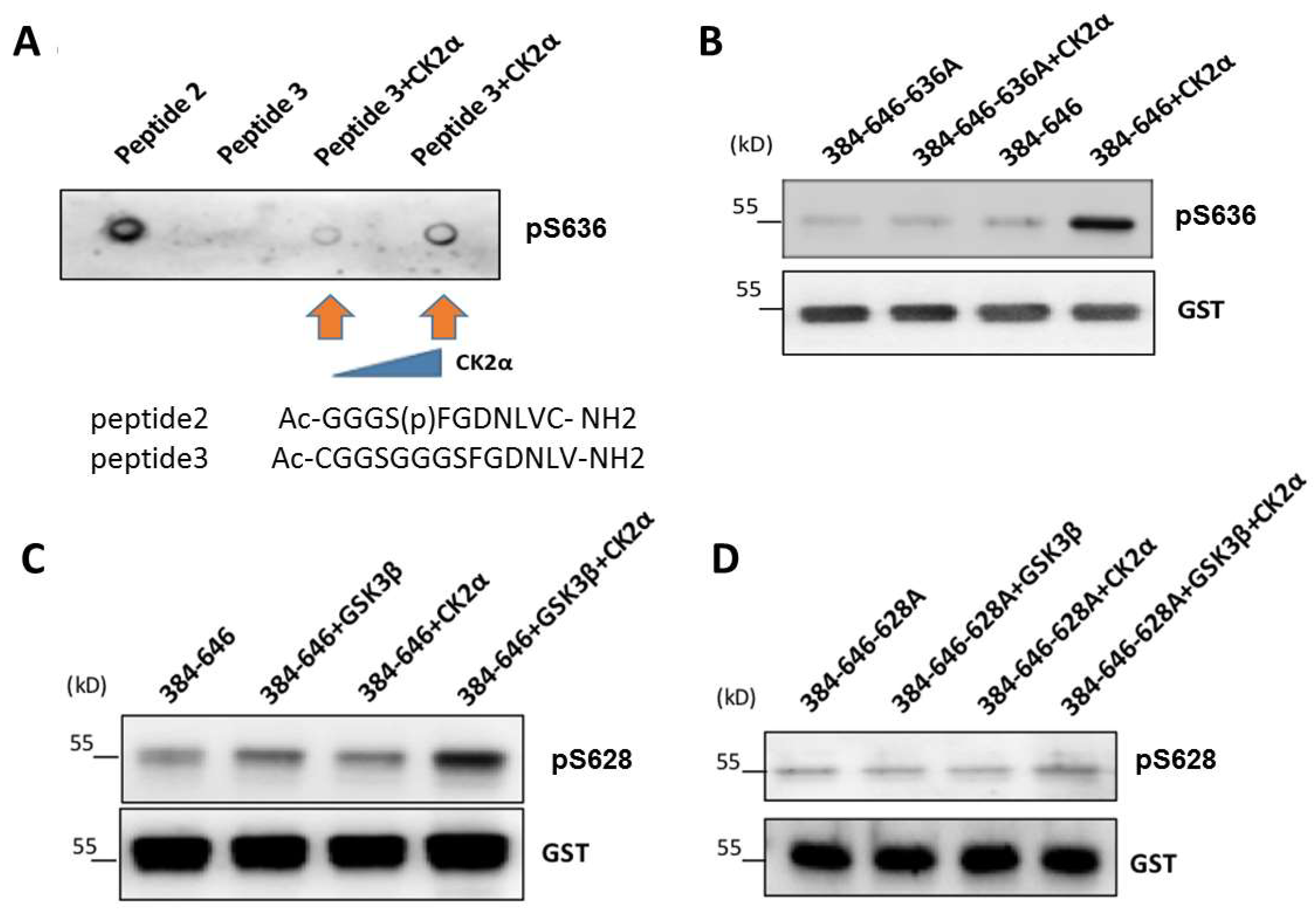
3.4. CK2-Induced Phosphorylation of S636 Triggers GSK3β-Induced Phosphorylation of Ser628
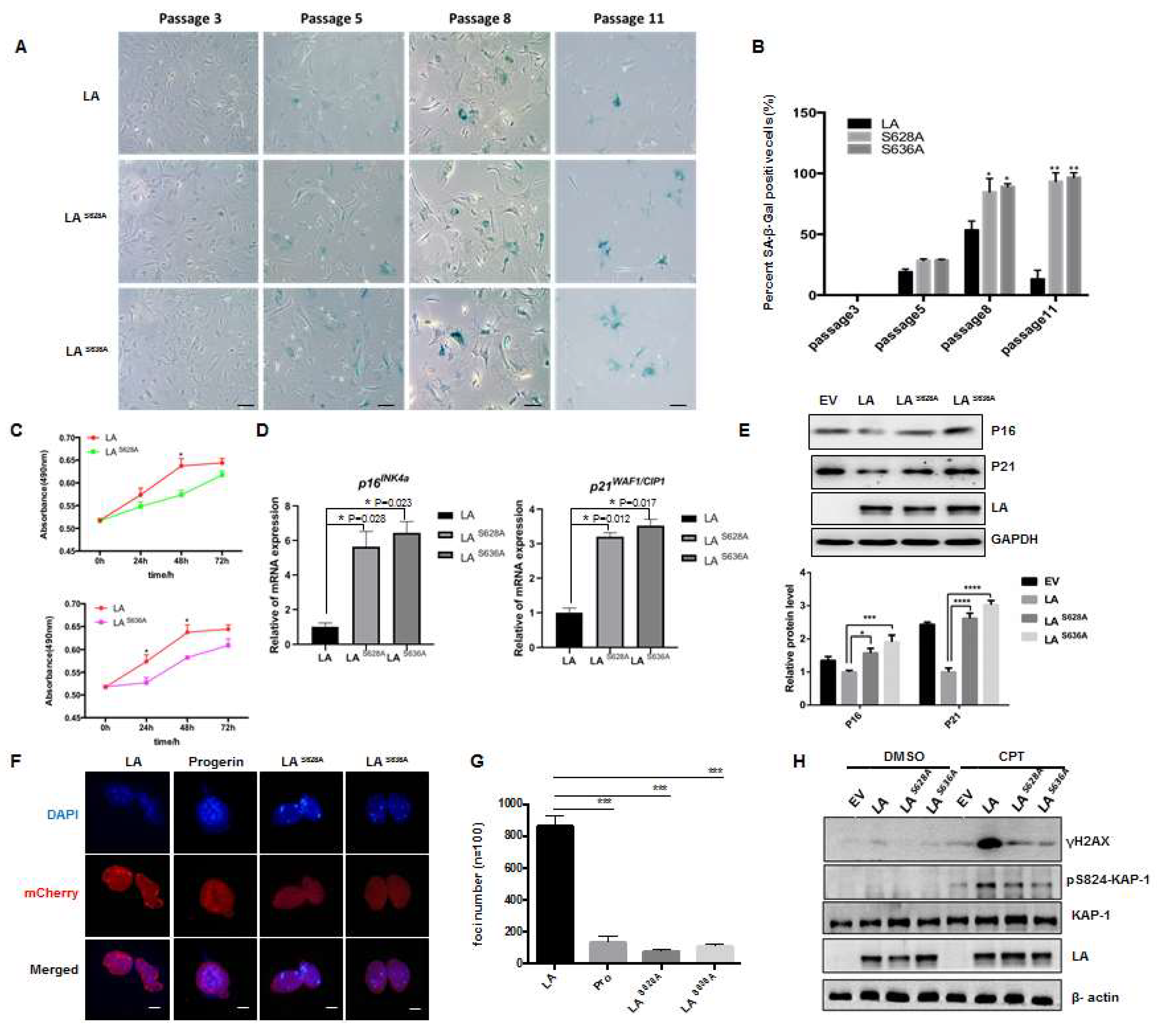
3.5. Role of Loss of Phosphorylation of Lamin A at Ser628 and Ser636 in Cellular Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peric-Hupkes, D.; van Steensel, B. Role of the Nuclear Lamina in Genome Organization and Gene Expression. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridger, J.; Foeger, N.; Kill, I.R.; Herrmann, H. The nuclear lamina. Both a structural framework and a platform for genome organization. FEBS J. 2007, 274, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Tramier, M.; Coppey-Moisan, M.; Gaillard, C.; Courvalin, J.-C.; Buendia, B. The truncated prelamin A in Hutchinson–Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum. Mol. Genet. 2006, 15, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Strelkov, S.V.; Schumacher, J.; Burkhard, P.; Aebi, U.; Herrmann, H. Crystal Structure of the Human Lamin A Coil 2B Dimer: Implications for the Head-to-tail Association of Nuclear Lamins. J. Mol. Biol. 2004, 343, 1067–1080. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [Green Version]
- Butin-Israeli, V.; Adam, S.A.; Goldman, A.E.; Goldman, R.D. Nuclear lamin functions and disease. Trends Genet. 2012, 28, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Graziotto, J.J.; Cao, K.; Collins, F.S.; Krainc, D. Rapamycin activates autophagy in Hutchinson-Gilford progeria syndrome: Implications for normal aging and age-dependent neurodegenerative disorders. Autophagy 2012, 8, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-Dependent Nuclear Defects in Human Aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranade, D.; Pradhan, R.; Jayakrishnan, M.; Hegde, S.; Sengupta, K. Lamin A/C and Emerin depletion impacts chromatin organization and dynamics in the interphase nucleus. BMC Cell Biol. 2019, 20, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaopa, M.A.; Ai, T.; Chao, B.; Xiao, X.; Vatta, M.; Habecker, B.A.; Ai, T. Phosphorylation of Lamin A/C at serine 22 modulates Na v 1.5 function. Physiol. Rep. 2021, 9, e15121. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-T.; Chen, Y.-H.; Chiu, W.-T.; Chen, H.-C. Tyrosine phosphorylation of lamin A by Src promotes disassembly of nuclear lamina in interphase. Life Sci. Alliance 2021, 4, e202101120. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Ikegami, K. Nuclear lamin phosphorylation: An emerging role in gene regulation and pathogenesis of laminopathies. Nucleus 2020, 11, 299–314. [Google Scholar] [CrossRef]
- Cho, S.; Abbas, A.; Irianto, J.; Ivanovska, I.L.; Xia, Y.; Tewari, M.; Discher, D.E. Progerin phosphorylation in interphase is lower and less mechanosensitive than lamin-A,C in iPS-derived mesenchymal stem cells. Nucleus 2018, 9, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.D.P.; Athirasala, A.; Kao, Y.-R.C.; Cho, S.; Harada, T.; Shin, J.-W.; et al. Matrix Elasticity Regulates Lamin-A,C Phosphorylation and Turnover with Feedback to Actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.; Goldman, A.; Pack, C.-G.; Melo-Cardenas, J.; Imanishi, S.; Goldman, R.D.; Eriksson, J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014, 127, 2683–2696. [Google Scholar] [CrossRef] [Green Version]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef]
- Ao, Y.; Zhang, J.; Liu, Z.; Qian, M.; Li, Y.; Wu, Z.; Sun, P.; Wu, J.; Bei, W.; Wen, J.; et al. Lamin A buffers CK2 kinase activity to modulate aging in a progeria mouse model. Sci. Adv. 2019, 5, eaav5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Qin, W.; Tang, X.; Meng, Y.; Hu, W.; Zhang, S.; Qian, M.; Liu, Z.; Cao, X.; Pang, Q.; et al. Vascular endothelium–targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci. Adv. 2020, 6, eaay5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Chen, B.; Liu, Z.; Gong, A.Y.; Gunning, W.T.; Ge, Y.; Malhotra, D.; Gohara, A.F.; Dworkin, L.D.; Gong, R. Age-related GSK3β overexpression drives podocyte senescence and glomerular aging. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Bali, S.K.; Bryce, D.; Prein, C.; Woodgett, J.R.; Beier, F. Glycogen synthase kinase 3 alpha/beta deletion induces precocious growth plate remodeling in mice. J. Mol. Med. 2021, 99, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Souder, D.C.; Anderson, R.M. An expanding GSK3 network: Implications for aging research. Geroscience 2019, 41, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.U.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, K.; Secchia, S.; Almakki, O.; Lieb, J.D.; Moskowitz, I.P. Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria. Dev. Cell 2020, 52, 699–713.e11. [Google Scholar] [CrossRef] [PubMed]
- Capanni, C.; Schena, E.; Di Giampietro, M.L.; Montecucco, A.; Mattioli, E.; Lattanzi, G. The role of prelamin A post-translational maturation in stress response and 53BP1 recruitment. Front. Cell Dev. Biol. 2022, 10, 1018102. [Google Scholar] [CrossRef]
- Liu, J.; Yin, X.; Liu, B.; Zheng, H.; Zhou, G.; Gong, L.; Li, M.; Li, X.; Wang, Y.; Hu, J.; et al. HP1α mediates defective heterochromatin repair and accelerates senescence inZmpste24-deficient cells. Cell Cycle 2014, 13, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wang, Z.; Ghosh, S.; Zhou, Z. Defective ATM-Kap-1-mediated chromatin remodeling impairs DNA repair and accelerates senescence in progeria mouse model. Aging Cell 2012, 12, 316–318. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Meta, M.; Coté, N.; Yang, S.H.; Stewart, C.L.; Sullivan, T.; Burghardt, A.; Majumdar, S.; Reue, K.; et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24 -deficient mice. Proc. Natl. Acad. Sci. USA 2004, 101, 18111–18116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, J.; Wang, B.; Brandenburg, S.M.; Basisty, N.; Evangelou, K.; Varela-Eirin, M.; Campisi, J.; Schilling, B.; Gorgoulis, V.; Demaria, M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 2021, 16, 2471–2498. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ao, Y.; Wu, Z.; Liao, Z.; Lan, J.; Zhang, J.; Sun, P.; Liu, B.; Wang, Z. Role of C-Terminal Phosphorylation of Lamin A in DNA Damage and Cellular Senescence. Cells 2023, 12, 639. https://doi.org/10.3390/cells12040639
Ao Y, Wu Z, Liao Z, Lan J, Zhang J, Sun P, Liu B, Wang Z. Role of C-Terminal Phosphorylation of Lamin A in DNA Damage and Cellular Senescence. Cells. 2023; 12(4):639. https://doi.org/10.3390/cells12040639
Chicago/Turabian StyleAo, Ying, Zhuping Wu, Zhiwei Liao, Juncong Lan, Jie Zhang, Pengfei Sun, Baohua Liu, and Zimei Wang. 2023. "Role of C-Terminal Phosphorylation of Lamin A in DNA Damage and Cellular Senescence" Cells 12, no. 4: 639. https://doi.org/10.3390/cells12040639