
Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity

 , , , , ,
, , , , ,
Abstract
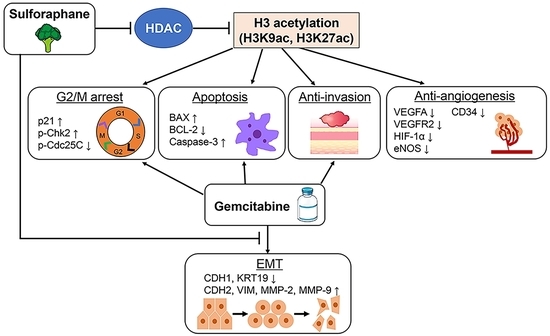
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Compounds and Cell Culture
2.2. Human iCCA Xenograft Model
2.3. Detection of HDAC/HAT Activity and Total Histone H3 and H4 Acetylation
2.4. Histone H3 Peptide Array
2.5. Cell Viability Assay and Analysis of Cytotoxic Synergy
2.6. Statistical Analysis
3. Results
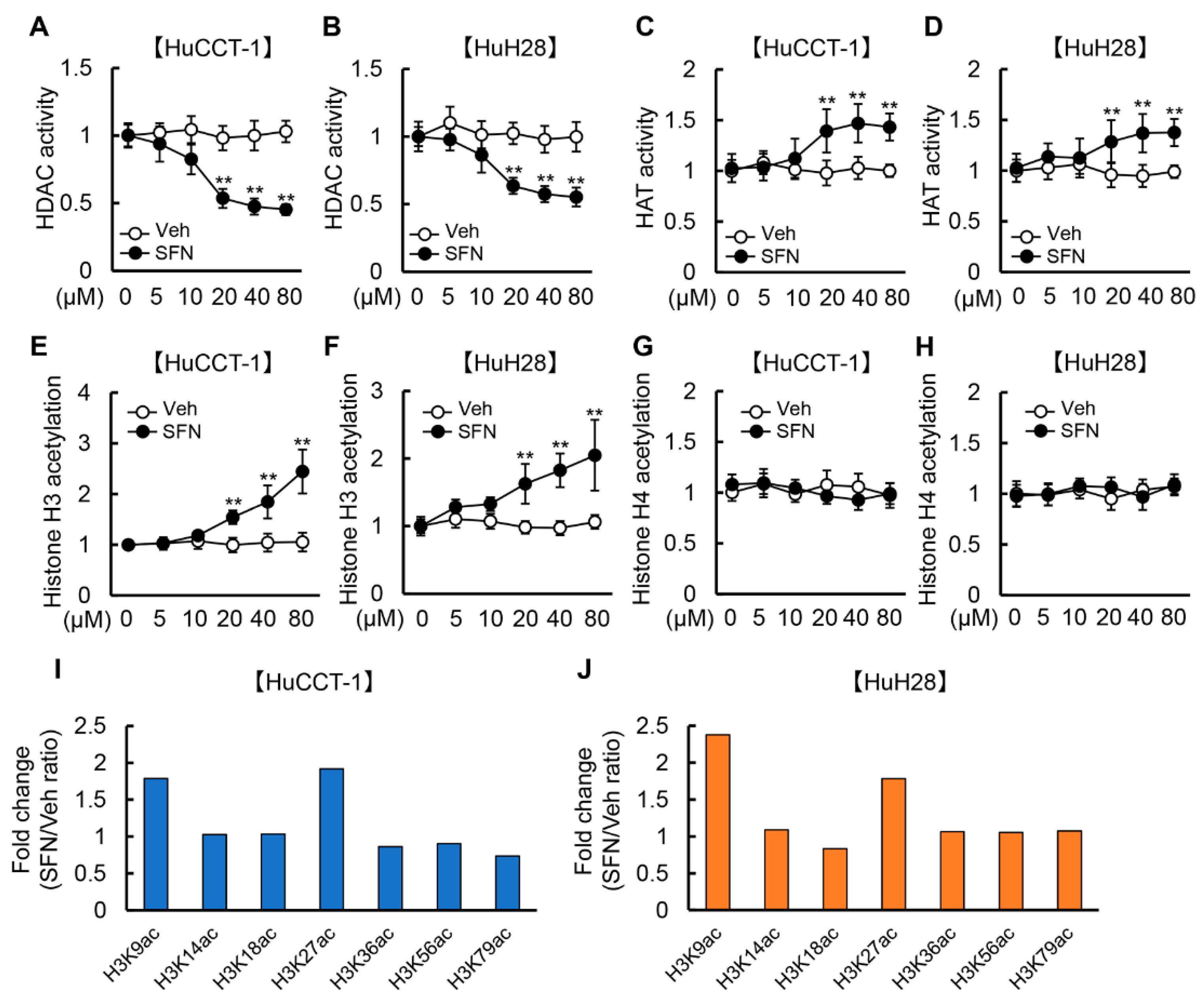
3.1. SFN Attenuates HDAC Activity and Promotes Histone H3 Acetylation in Human iCCA Cells
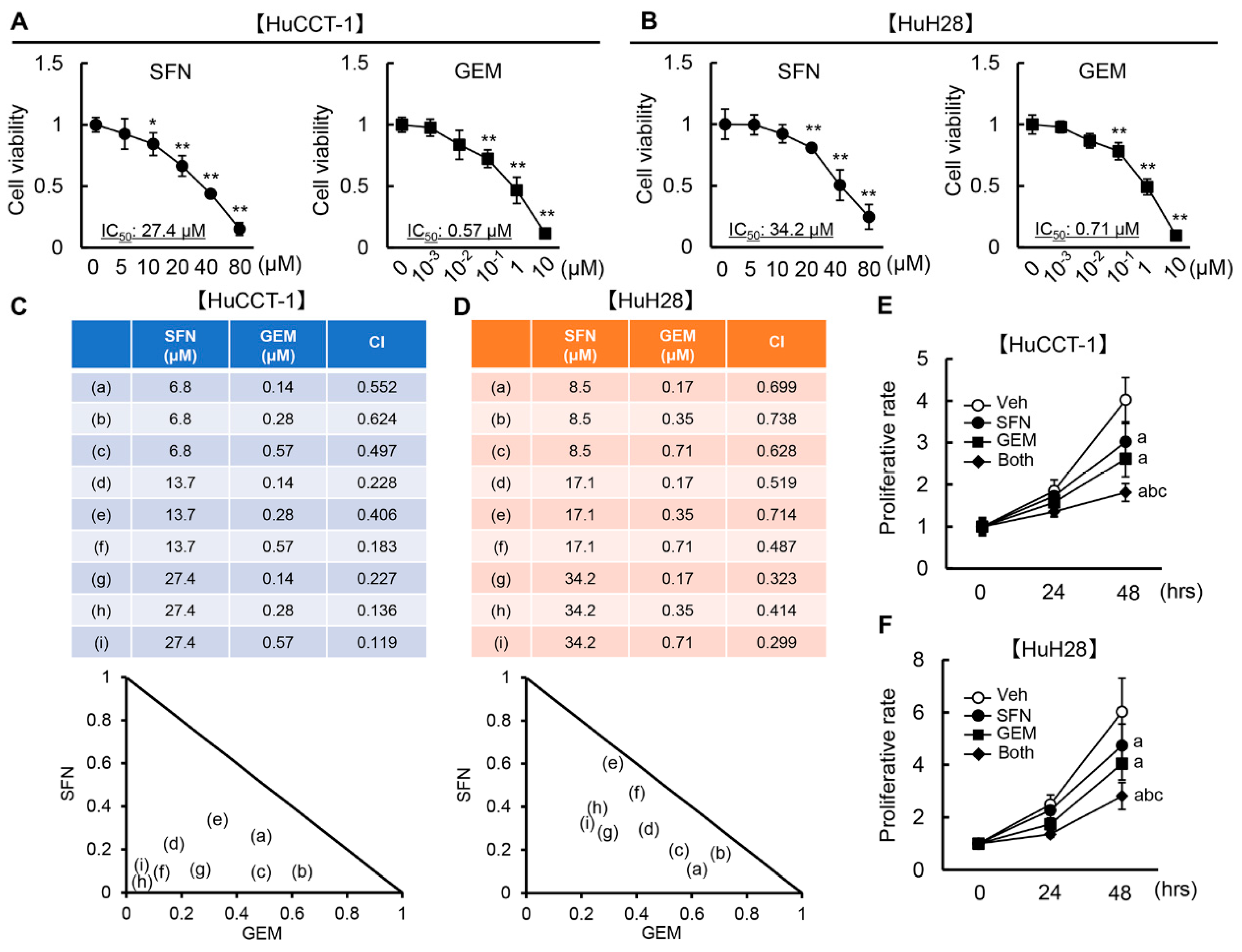
3.2. SFN Has a Synergistic Effect with GEM-Mediated Cell Growth Inhibition in Human iCCA Cells
3.3. SFN Induces G2/M Arrest and Promotes Apoptosis in Human iCCA Cells
3.4. SFN Inhibits Cancer Cell Invasion, Migration, Angiogenic Activity, and Epithelial-Mesenchymal Transition (EMT) in Human iCCA Cells
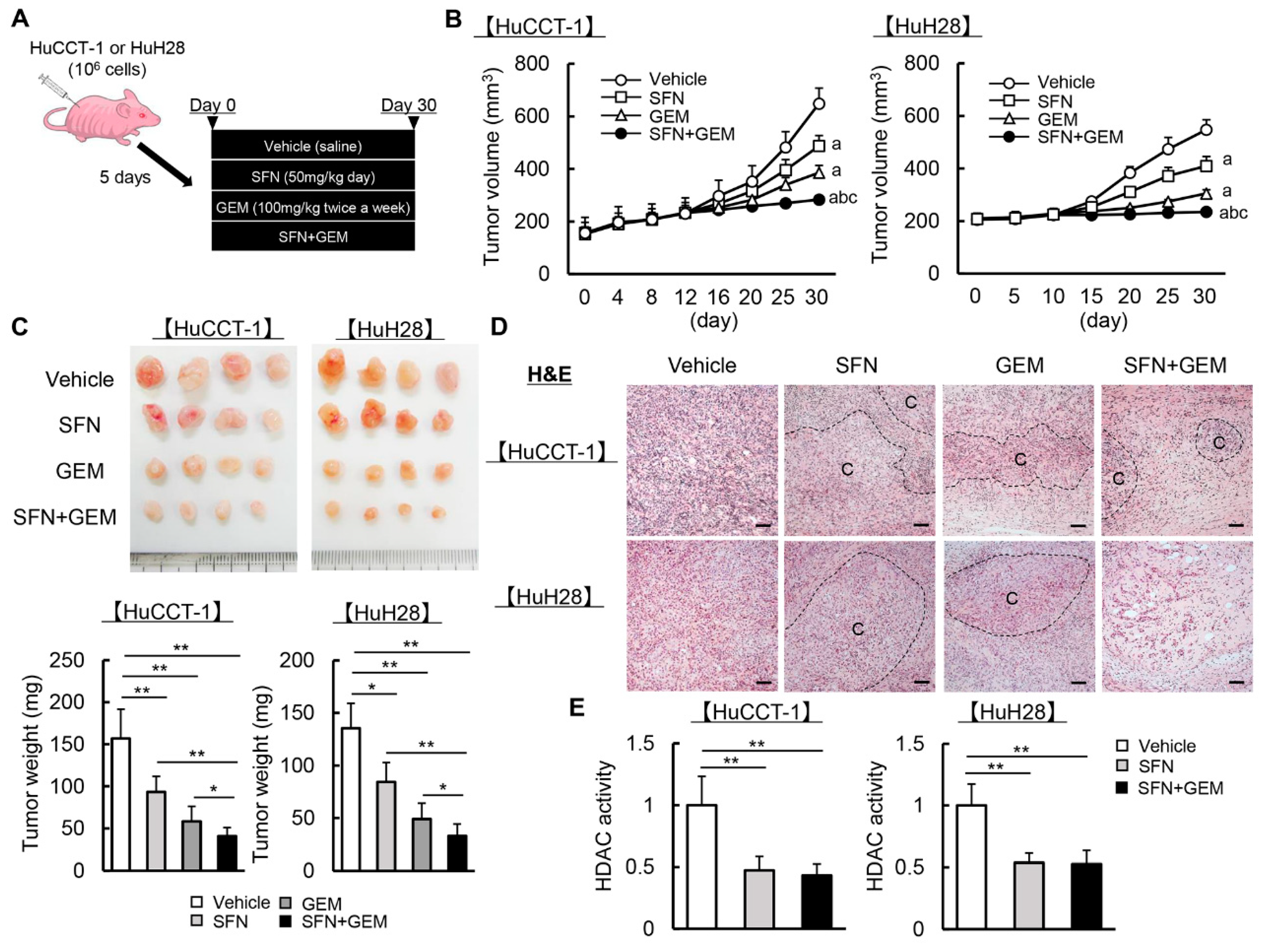
3.5. SFN Potentiates the GEM-Mediated Reduction of the Human iCCA-Derived Xenograft Tumor Growth
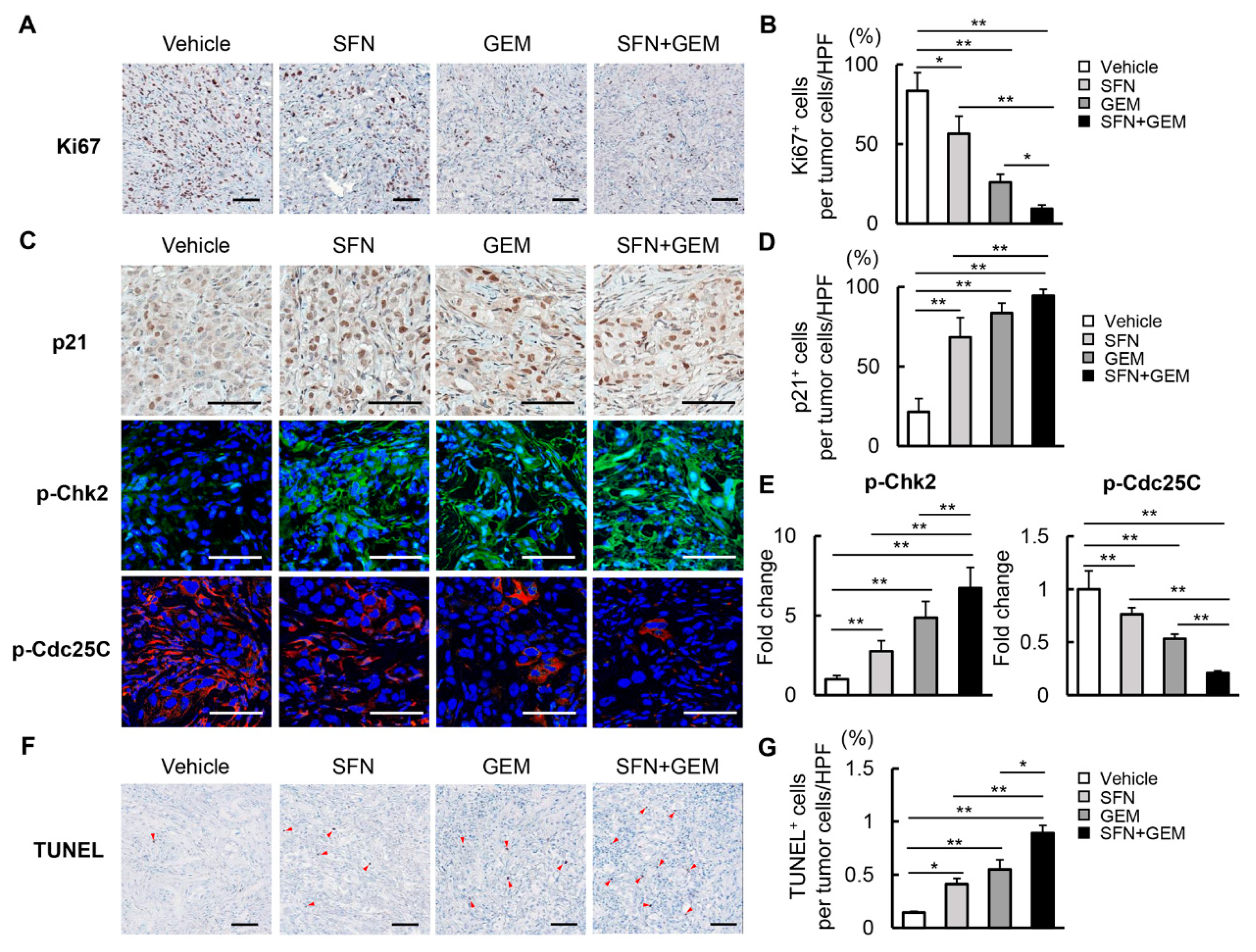
3.6. SFN Suppresses Cell Proliferation and Induces Apoptosis in Human iCCA-Derived Xenograft Tumors
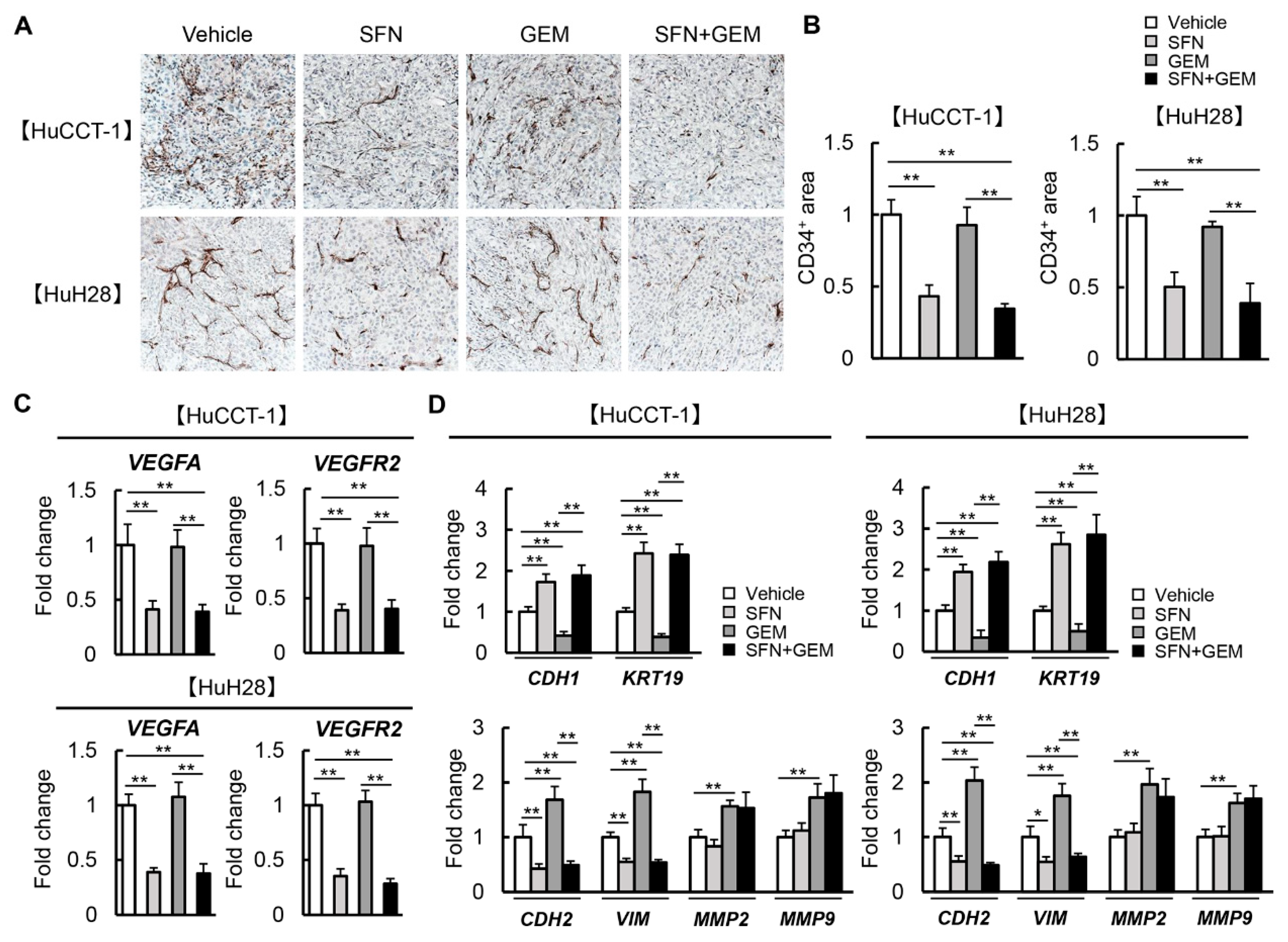
3.7. SFN Attenuates Intratumor Angiogenesis and GEM-Mediated EMT in Human iCCA-Derived Xenograft Tumors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Ivenson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Petri, G.L.; Pecoraro, C.; Randazzo, O.; Zoppi, S.; Cascioferro, S.M.; Parrino, B.; Carbone, D.; Hassouni, B.E.; Cavazzoni, A.; Zaffaroni, N.; et al. New Imidazo[2,1-b][1,3,4]Thiadiazole Derivatives Inhibit FAK Phosphorylation and Potentiate the Antiproliferative Effects of Gemcitabine Through Modulation of the Human Equilibrative Nucleoside Transporter-1 in Peritoneal Mesothelioma. Anticancer Res. 2020, 40, 4913–4919. [Google Scholar] [CrossRef]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing effective combination therapy for pancreatic cancer: An overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef]
- Merarchi, M.; Sethi, G.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Ahn, K.S. Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules 2019, 24, 1047. [Google Scholar] [CrossRef] [Green Version]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 15, 277. [Google Scholar] [CrossRef] [PubMed]
- Prter, N.J.; Christianson, D.W. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Xie, H.J.; Chang, Y.G.; Kim, M.G.; Park, H.; Lee, J.Y.; et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J. Cell. Biochem. 2012, 113, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Hu, H.; Yuan, C.; Jin, Z.; Guo, Z.; Wang, L.; Wang, L. Histone deacetylase 3 promotes pancreatic cancer cell proliferation, invasion and increases drug-resistance through histone modification of P27, P53 and Bax. Int. J. Oncol. 2014, 45, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Morine, Y.; Shimada, M.; Iwahashi, S.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Miyake, H. Role of histone deacetylase expression in intrahepatic cholangiocarcinoma. Surgery 2012, 151, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, J.; Huo, Q.; Chen, Z.; Chen, J.; Xu, X. Chidamide Suppresses the Growth of Cholangiocarcinoma by Inhibiting HDAC3 and Promoting FOXO1 Acetylation. Stem. Cells Int. 2022, 2022, 3632549. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Li, X.; Wang, Y.; Lu, Y. The protective effect of sulforaphane on type II diabetes induced by high-fat diet and low-dosage streptozotocin. Food Sci. Nutr. 2020, 9, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Romeo, L.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. Isothiocyanates: An Overview of Their Antimicrobial Activity against Human Infections. Molecules 2018, 23, 624. [Google Scholar] [CrossRef] [Green Version]
- Tortorella, S.M.; Royce, S.G.; Licciardi, P.V.; Karagiannis, T.C. Dietary Sulforaphane in Cancer Chemoprevention: The Role of Epigenetic Regulation and HDAC Inhibition. Antioxid. Redox Signal. 2015, 22, 1382–1424. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Jiang, X.; Meng, L.; Dong, X.; Shen, Y.; Xin, Y. Anticancer Activity of Sulforaphane: The Epigenetic Mechanisms and the Nrf2 Signaling Pathway. Oxid. Med. Cell Longev. 2018, 2018, 5438179. [Google Scholar] [CrossRef] [Green Version]
- Rackauskas, R.; Zhou, D.; Uselis, S.; Strupas, K.; Herr, I.; Schemmer, P. Sulforaphane sensitizes human cholangiocarcinoma to cisplatin via the downregulation of anti-apoptotic proteins. Oncol. Rep. 2017, 37, 3660–3666. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Park, H.J.; Moon, D.O. Sulforaphane sensitizes human breast cancer cells to paclitaxel-induced apoptosis by downregulating the NF-κB signaling pathway. Oncol. Lett. 2017, 13, 4427–4432. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, K.; Moriya, K.; Kaji, K.; Saikawa, S.; Sato, S.; Nishimura, N.; Namisaki, T.; Akahane, T.; Mitoro, A.; Yoshiji, H. Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2020, 21, 7588. [Google Scholar] [CrossRef]
- Rai, R.; Essel, K.G.; Benbrook, D.M.; Garland, J.; Zhao, Y.D.; Chandra, V. Preclinical Efficacy and Involvement of AKT, mTOR, and ERK Kinases in the Mechanism of Sulforaphane against Endometrial Cancer. Cancers 2020, 12, 1273. [Google Scholar] [CrossRef]
- Mathison, A.; de Assuncao, T.M.; Dsouza, N.R.; Williams, M.; Zimmermann, M.T.; Urrutia, R.; Lomberk, G. Discovery, expression, cellular localization, and molecular properties of a novel, alternative spliced HP1γ isoform, lacking the chromoshadow domain. PLoS ONE 2020, 15, e0217452. [Google Scholar] [CrossRef] [Green Version]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [Green Version]
- Mino, M.; Kanno, K.; Okimoto, K.; Sugiyama, A.; Kishikawa, N.; Kobayashi, T.; Ono, J.; Izuhara, K.; Kobayashi, T.; Ohigashi, T.; et al. Periostin promotes malignant potential by induction of epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatol. Commnu. 2017, 1, 1099–1109. [Google Scholar] [CrossRef]
- Yin, Y.; Zhang, M.; Dorfman, R.G.; Li, Y.; Zhao, Z.; Pan, Y.; Zhou, Q.; Huang, S.; Zhao, S.; Yao, Y.; et al. Histone deacetylase 3 overexpression in human cholangiocarcinoma and promotion of cell growth via apoptosis inhibition. Cell Death Dis. 2017, 8, e2856. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; Larusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Lee, E.J.; Ji, M.; Park, S.M. HDAC inhibitors, trichostatin A and valproic acid, increase E-cadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol. Rep. 2018, 40, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Saenglee, S.; Senawong, G.; Jogloy, S.; Sripa, B.; Sanewong, T. Peanut testa extracts possessing histone deacetylase inhibitory activity induce apoptosis in cholangiocarcinoma cells. Biomed. Pharmacother. 2018, 98, 233–241. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Trafails, D.T.; Franco, R.; Zoumpourlis, V.; Pappa, A.; Panayiotidis, M.I. Sulforaphane and iberin are potent epigenetic modulators of histone acetylation and methylation in malignant melanoma. Eur. J. Nutr. 2021, 60, 147–158. [Google Scholar] [CrossRef]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Qin, X.; Zhou, Y.; Li, G.; Liu, Z.; Geng, X.; Yue, H. Long non-coding RNA LINC00665 promotes gemcitabine resistance of Cholangiocarcinoma cells via regulating EMT and stemness properties through miR-424-5p/BCL9L axis. Cell Death Dis. 2021, 12, 72. [Google Scholar] [CrossRef]
- Tang, H.M.; Kuay, K.T.; Koh, P.F.; Asad, M.; Tan, T.Z.; Chung, V.Y.; Lee, S.C.; Thiery, J.P.; Huang, R.J. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.X.; Zou, Y.J.; Zhuang, X.B.; Chen, S.X.; Lin, Y.; Li, W.L.; Lin, J.J.; Lin, Z.Q. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3β/β-catenin signaling pathways. Acta Pharmacol. Sin. 2017, 38, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, M.; Fazli, M.; Saeednia, S.; Kharanagh, M.G.; Ahmadiankia, N. Sulforaphane Modulates Cell Migration and Expression of β-Catenin and Epithelial Mesenchymal Transition Markers in Breast Cancer Cells. Iran. J. Public Health 2020, 49, 77–85. [Google Scholar]
- Wu, J.; Han, J.; Hou, B.; Deng, C.; Wu, H.; Shen, L. Sulforaphane inhibits TGF-β-induced epithelial-mesenchymal transition of hepatocellular carcinoma cells via the reactive oxygen species-dependent pathway. Oncol. Rep. 2016, 35, 2977–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, J.Q.; Ge, M.M.; Zhang, Q.; Wang, X.Q.; Zhu, J.Y.; Xie, C.F.; Li, X.T.; Zhong, C.Y.; Han, H.Y. Sulforaphane inhibits epithelial-mesenchymal transition by activating extracellular signal-regulated kinase 5 in lung cancer cells. J. Nutr. Biochem. 2019, 72, 108219. [Google Scholar] [CrossRef]
- Li, S.H.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol. Cell. Biochem. 2013, 373, 217–227. [Google Scholar] [CrossRef]
- Cai, C.; Wang, X.; Fu, Q.; Chen, A. The VEGF expression associated with prognosis in patients with intrahepatic cholangiocarcinoma: A systematic review and meta-analysis. World J. Surg. Oncol. 2022, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, H.; Zhang, Z.; Jiang, B.H.; Luo, J.; Shi, X. Sulforaphane inhibited expression of hypoxia-inducible factor-1alpha in human tongue squamous cancer cells and prostate cancer cells. Int. J. Cancer 2008, 123, 1255–1261. [Google Scholar] [CrossRef]
- Kim, D.H.; Sung, B.; Kang, Y.J.; Hwang, S.Y.; Kim, M.J.; Yoon, J.H.; Im, E.; Kim, N.D. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int. J. Oncol. 2015, 47, 2226–2232. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Atkinson, S.J.; Akbareian, S.E.; Zhou, Z.; Munsterberg, A.; Robinson, S.D.; Bao, Y. Sulforaphane exerts anti-angiogenesis effects against hepatocellular carcinoma through inhibition of STAT3/HIF-1α/VEGF signalling. Sci. Rep. 2017, 7, 12651. [Google Scholar] [CrossRef] [Green Version]
- Bertl, E.; Bartsch, H.; Gerhauser, C. Inhibition of angiogenesis and endothelial cell functions are novel sulforaphane-mediated mechanisms in chemoprevention. Mol. Cancer Ther. 2006, 5, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Kunnumakkara, A.B.; Guha, S.; Krishnan, S.; Diagaradjane, P.; Gelovani, J.; Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007, 67, 3853–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitch, A.D.; Law, H.; White, R.D. A stable propidium iodide staining procedure for flow cytometry. J. Histochem. Cytochem. 1982, 30, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomooka, F.; Kaji, K.; Nishimura, N.; Kubo, T.; Iwai, S.; Shibamoto, A.; Suzuki, J.; Kitagawa, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity. Cells 2023, 12, 687. https://doi.org/10.3390/cells12050687
Tomooka F, Kaji K, Nishimura N, Kubo T, Iwai S, Shibamoto A, Suzuki J, Kitagawa K, Namisaki T, Akahane T, et al. Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity. Cells. 2023; 12(5):687. https://doi.org/10.3390/cells12050687
Chicago/Turabian StyleTomooka, Fumimasa, Kosuke Kaji, Norihisa Nishimura, Takahiro Kubo, Satoshi Iwai, Akihiko Shibamoto, Junya Suzuki, Koh Kitagawa, Tadashi Namisaki, Takemi Akahane, and et al. 2023. "Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity" Cells 12, no. 5: 687. https://doi.org/10.3390/cells12050687
APA StyleTomooka, F., Kaji, K., Nishimura, N., Kubo, T., Iwai, S., Shibamoto, A., Suzuki, J., Kitagawa, K., Namisaki, T., Akahane, T., Mitoro, A., & Yoshiji, H. (2023). Sulforaphane Potentiates Gemcitabine-Mediated Anti-Cancer Effects against Intrahepatic Cholangiocarcinoma by Inhibiting HDAC Activity. Cells, 12(5), 687. https://doi.org/10.3390/cells12050687