
Stem Cell Factor and Granulocyte Colony-Stimulating Factor Promote Remyelination in the Chronic Phase of Severe Traumatic Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Controlled Cortical Impact Model of TBI
2.2. Experimental Design
2.3. Immunofluorescence Staining
2.4. Quantification of Immunofluorescence Staining
2.5. Culture of Oligodendrocyte Progenitor Cells Isolated from Adult Mouse Brain
2.6. Proliferation and Differentiation Assay of Oligodendrocyte Progenitor Cells
2.7. Statistical Analysis
3. Results
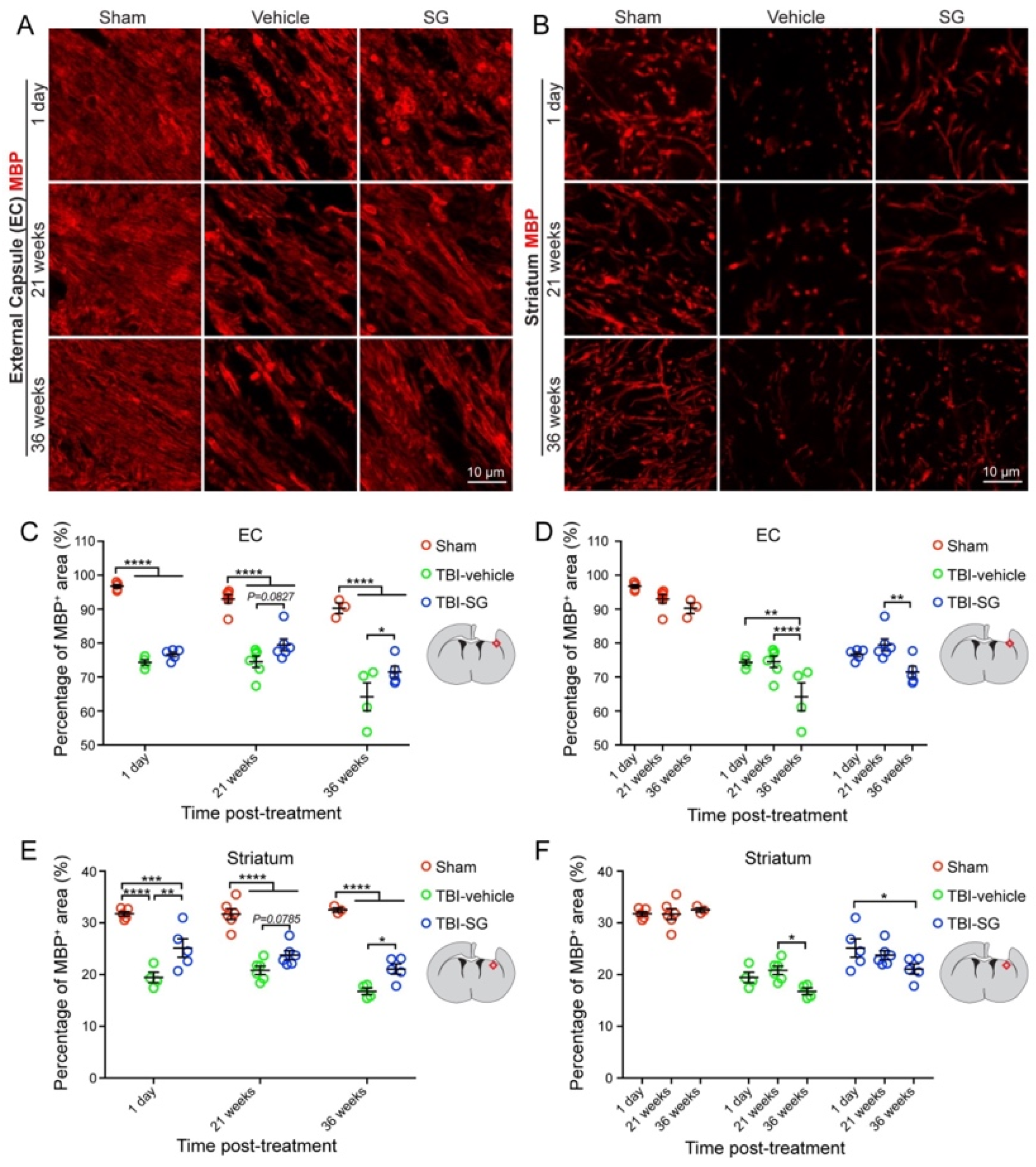
3.1. SCF + G-CSF Treatment Increases Remyelination in the Chronic Phase of Severe TBI
3.2. SCF + G-CSF Treatment Increases Olig2 Positive Cells in the Ipsilateral External Capsule in the Chronic Phase of Severe TBI
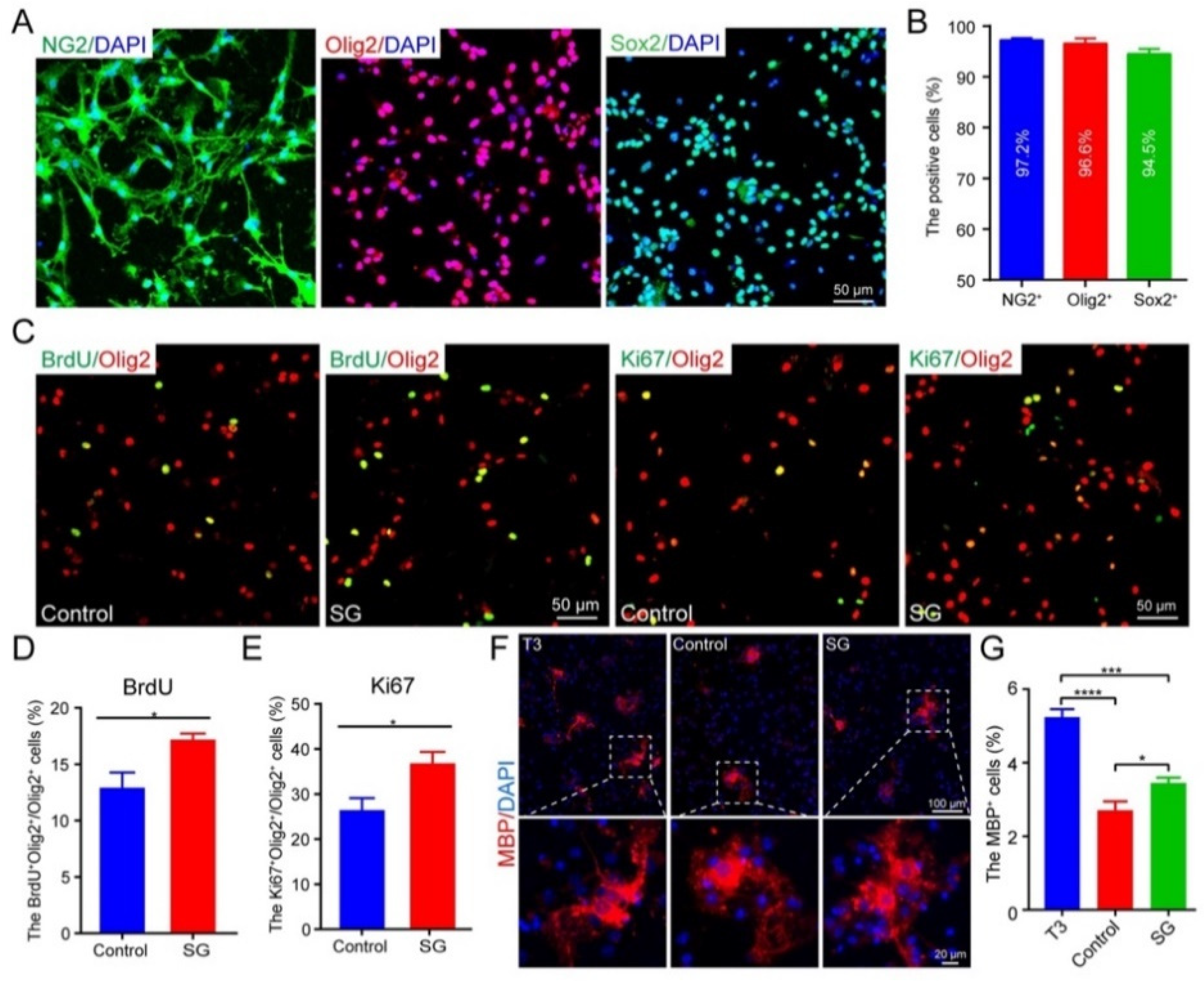
3.3. SCF + G-CSF Treatment Increases Oligodendrocyte Progenitor Cells in the Subventricular Zone in the Chronic Phase of Severe TBI
3.4. SCF + G-CSF Treatment Promotes Oligodendrocyte Progenitor Cell Proliferation and Differentiation In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armstrong, R.C.; Mierzwa, A.J.; Sullivan, G.M.; Sanchez, M.A. Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 2016, 110, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury–Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR. Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. TBI Data. 2022. Available online: https://www.cdc.gov/traumaticbraininjury/data/tbi-edhd.html (accessed on 1 February 2023).
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Ng, H.K.; Mahaliyana, R.D.; Poon, W.S. The pathological spectrum of diffuse axonal injury in blunt head trauma: Assessment with axon and myelin strains. Clin. Neurol. Neurosurg. 1994, 96, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Adnan, A.; Crawley, A.; Mikulis, D.; Moscovitch, M.; Colella, B.; Green, R. Moderate-severe traumatic brain injury causes delayed loss of white matter integrity: Evidence of fornix deterioration in the chronic stage of injury. Brain Inj. 2013, 27, 1415–1422. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.C.; Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp. Neurol. 2016, 275 Pt 3, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spader, H.S.; Dean, D.C.; LaFrance, W.C.; Raukar, N.P.; Cosgrove, G.R.; Eyerly-Webb, S.A.; Ellermeier, A.; Correia, S.; Deoni, S.C.L.; Rogg, J. Prospective study of myelin water fraction changes after mild traumatic brain injury in collegiate contact sports. J. Neurosurg. 2019, 130, 1321–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Xu, J.; Zhan, T.; Wang, H.; Huang, X.; Liu, W.; Yang, X.; Zhan, R. The occurrence of diffuse axonal injury in the brain: Associated with the accumulation and clearance of myelin debris. Neural Regen. Res. 2014, 9, 1902–1906. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filbin, M.T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003, 4, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flygt, J.; Djupsjo, A.; Lenne, F.; Marklund, N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur. J. Neurosci. 2013, 38, 2153–2165. [Google Scholar] [CrossRef]
- Dent, K.A.; Christie, K.J.; Bye, N.; Basrai, H.S.; Turbic, A.; Habgood, M.; Cate, H.S.; Turnley, A.M. Oligodendrocyte birth and death following traumatic brain injury in adult mice. PLoS ONE 2015, 10, e0121541. [Google Scholar] [CrossRef] [Green Version]
- Takase, H.; Washida, K.; Hayakawa, K.; Arai, K.; Wang, X.; Lo, E.H.; Lok, J. Oligodendrogenesis after traumatic brain injury. Behav. Brain Res. 2018, 340, 205–211. [Google Scholar] [CrossRef]
- Marion, C.M.; Radomski, K.L.; Cramer, N.P.; Galdzicki, Z.; Armstrong, R.C. Experimental Traumatic Brain Injury Identifies Distinct Early and Late Phase Axonal Conduction Deficits of White Matter Pathophysiology, and Reveals Intervening Recovery. J. Neurosci. 2018, 38, 8723–8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, S.J.; Zsebo, K.M.; Geissler, E.N. The kit ligand, stem cell factor. Adv. Immunol 1994, 55, 1–96. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Griffin, J.D. Granulocyte colony-stimulating factor and its receptor. Blood 1991, 78, 2791–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.R.; Navalitloha, Y.; Singhal, S.; Mehta, J.; Piao, C.S.; Guo, W.P.; Kessler, J.A.; Groothuis, D.R. Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Exp. Neurol. 2007, 204, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Kruger, C.; Steigleder, T.; Weber, D.; Pitzer, C.; Laage, R.; Aronowski, J.; Maurer, M.H.; Gassler, N.; Mier, W.; et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Investig. 2005, 115, 2083–2098. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, S.J.; Sinn, D.I.; Kim, S.U.; Kim, M.; Roh, J.K. Granulocyte colony-stimulating factor stimulates neurogenesis via vascular endothelial growth factor with STAT activation. Brain Res. 2006, 1073–1074, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Toshkezi, G.; Kyle, M.; Longo, S.L.; Chin, L.S.; Zhao, L.R. Brain repair by hematopoietic growth factors in the subacute phase of traumatic brain injury. J. Neurosurg. 2018, 129, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhao, L.R. Turning Death to Growth: Hematopoietic Growth Factors Promote Neurite Outgrowth through MEK/ERK/p53 Pathway. Mol. Neurobiol. 2018, 55, 5913–5925. [Google Scholar] [CrossRef] [PubMed]
- Kadota, R.; Koda, M.; Kawabe, J.; Hashimoto, M.; Nishio, Y.; Mannoji, C.; Miyashita, T.; Furuya, T.; Okawa, A.; Takahashi, K.; et al. Granulocyte colony-stimulating factor (G-CSF) protects oligodendrocyte and promotes hindlimb functional recovery after spinal cord injury in rats. PLoS ONE 2012, 7, e50391. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Watanabe, M.; Hasuo, A.; Imai, M.; Suyama, K.; Sakai, D.; Kawada, H.; Matsumae, M.; Mochida, J. Efficacy of the coadministration of granulocyte colony-stimulating factor and stem cell factor in the activation of intrinsic cells after spinal cord injury in mice. J. Neurosurg. Spine 2010, 13, 516–523. [Google Scholar] [CrossRef]
- Qiu, X.; Ping, S.; Kyle, M.; Chin, L.; Zhao, L.R. Long-term beneficial effects of hematopoietic growth factors on brain repair in the chronic phase of severe traumatic brain injury. Exp. Neurol. 2020, 330, 113335. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485.e478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, E.M.; May, G.J.; Nelson, S.M. MRI-based measures of intracortical myelin are sensitive to a history of TBI and are associated with functional connectivity. Neuroimage 2019, 200, 199–209. [Google Scholar] [CrossRef]
- Herrera, J.J.; Bockhorst, K.; Kondraganti, S.; Stertz, L.; Quevedo, J.; Narayana, P.A. Acute White Matter Tract Damage after Frontal Mild Traumatic Brain Injury. J. Neurotrauma 2017, 34, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, H.; Nabeshima, Y.; Yoshida, S.; Chisaka, O.; Ikenaka, K.; Nabeshima, Y. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr. Biol. 2002, 12, 1157–1163. [Google Scholar] [CrossRef] [Green Version]
- Jakovcevski, I.; Zecevic, N. Olig transcription factors are expressed in oligodendrocyte and neuronal cells in human fetal CNS. J. Neurosci. 2005, 25, 10064–10073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stassart, R.M.; Mobius, W.; Nave, K.A.; Edgar, J.M. The Axon-Myelin Unit in Development and Degenerative Disease. Front. Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, G.M.; Mierzwa, A.J.; Kijpaisalratana, N.; Tang, H.; Wang, Y.; Song, S.K.; Selwyn, R.; Armstrong, R.C. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J. Neuropathol. Exp. Neurol. 2013, 72, 1106–1125. [Google Scholar] [CrossRef] [Green Version]
- Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M.; McDaniel, D.P.; Armstrong, R.C. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 2015, 74, 218–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, H.; Ghosh, P.; Kernie, S.G. Subventricular zone neural stem cells remodel the brain following traumatic injury in adult mice. J. Neurotrauma 2004, 21, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Adorjan, I.; Mundim, M.V.; Sun, B.; Dizon, M.L.; Szele, F.G. Traumatic Brain Injury Activation of the Adult Subventricular Zone Neurogenic Niche. Front. Neurosci. 2016, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazel, C.Y.; Limke, T.L.; Osborne, J.K.; Miura, T.; Cai, J.; Pevny, L.; Rao, M.S. Sox2 expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell 2005, 4, 197–207. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, X.; Gui, X.; Croteau, C.; Song, L.; Xu, J.; Wang, A.; Bannerman, P.; Guo, F. Sox2 Is Essential for Oligodendroglial Proliferation and Differentiation during Postnatal Brain Myelination and CNS Remyelination. J. Neurosci. 2018, 38, 1802–1820. [Google Scholar] [CrossRef] [Green Version]
- Kinnunen, K.M.; Greenwood, R.; Powell, J.H.; Leech, R.; Hawkins, P.C.; Bonnelle, V.; Patel, M.C.; Counsell, S.J.; Sharp, D.J. White matter damage and cognitive impairment after traumatic brain injury. Brain 2011, 134, 449–463. [Google Scholar] [CrossRef]
- Filley, C.M.; Kelly, J.P. White Matter and Cognition in Traumatic Brain Injury. J. Alzheimers Dis. 2018, 65, 345–362. [Google Scholar] [CrossRef]
- Kraus, M.F.; Susmaras, T.; Caughlin, B.P.; Walker, C.J.; Sweeney, J.A.; Little, D.M. White matter integrity and cognition in chronic traumatic brain injury: A diffusion tensor imaging study. Brain 2007, 130, 2508–2519. [Google Scholar] [CrossRef]
- Narayana, P.A. White matter changes in patients with mild traumatic brain injury: MRI perspective. Concussion 2017, 2, CNC35. [Google Scholar] [CrossRef] [Green Version]
- Abdel Baki, S.G.; Schwab, B.; Haber, M.; Fenton, A.A.; Bergold, P.J. Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS ONE 2010, 5, e12490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, M.; James, J.; Kim, J.; Sangobowale, M.; Irizarry, R.; Ho, J.; Nikulina, E.; Grin’kina, N.M.; Ramadani, A.; Hartman, I.; et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Flygt, J.; Gumucio, A.; Ingelsson, M.; Skoglund, K.; Holm, J.; Alafuzoff, I.; Marklund, N. Human Traumatic Brain Injury Results in Oligodendrocyte Death and Increases the Number of Oligodendrocyte Progenitor Cells. J. Neuropathol. Exp. Neurol. 2016, 75, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [Green Version]
- Pepper, R.E.; Pitman, K.A.; Cullen, C.L.; Young, K.M. How Do Cells of the Oligodendrocyte Lineage Affect Neuronal Circuits to Influence Motor Function, Memory and Mood? Front. Cell. Neurosci. 2018, 12, 399. [Google Scholar] [CrossRef] [Green Version]
- Mitew, S.; Xing, Y.L.; Merson, T.D. Axonal activity-dependent myelination in development: Insights for myelin repair. J. Chem. Neuroanat. 2016, 76, 2–8. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Mason, J.L.; Toews, A.; Hostettler, J.D.; Morell, P.; Suzuki, K.; Goldman, J.E.; Matsushima, G.K. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am. J. Pathol. 2004, 164, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, N.; Pham, L.D.; Hayakawa, K.; Matsuzaki, T.; Seo, J.H.; Magnain, C.; Ayata, C.; Kim, K.W.; Boas, D.; Lo, E.H.; et al. Age-related decline in oligodendrogenesis retards white matter repair in mice. Stroke 2013, 44, 2573–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, T.; Liang, A.C.; Miyamoto, N.; Lo, E.H.; Arai, K. Mechanisms of oligodendrocyte regeneration from ventricular-subventricular zone-derived progenitor cells in white matter diseases. Front. Cell. Neurosci. 2013, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Abe, K. Endogenous neurogenesis, oligodendrogenesis and angiogenesis after ischemic brain injury. J. Neurol. Neurophysiol. 2012, s8, 1–4. [Google Scholar] [CrossRef]
- Ida, J.A., Jr.; Dubois-Dalcq, M.; McKinnon, R.D. Expression of the receptor tyrosine kinase c-kit in oligodendrocyte progenitor cells. J. Neurosci. Res. 1993, 36, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Cui, L.; Piao, C.; Li, B.; Zhao, L.R. The effects of hematopoietic growth factors on neurite outgrowth. PLoS ONE 2013, 8, e75562. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.L.; Wood, R.J.; Nguyen, J.; Norman, E.M.L.; Jun, C.M.K.; Prawdiuk, A.R.; Biemond, M.; Nguyen, H.T.H.; Northfield, S.E.; Hughes, R.A.; et al. Targeting TrkB with a Brain-Derived Neurotrophic Factor Mimetic Promotes Myelin Repair in the Brain. J. Neurosci. 2018, 38, 7088–7099. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, X.; Ping, S.; Kyle, M.; Chin, L.; Zhao, L.-R. Stem Cell Factor and Granulocyte Colony-Stimulating Factor Promote Remyelination in the Chronic Phase of Severe Traumatic Brain Injury. Cells 2023, 12, 705. https://doi.org/10.3390/cells12050705
Qiu X, Ping S, Kyle M, Chin L, Zhao L-R. Stem Cell Factor and Granulocyte Colony-Stimulating Factor Promote Remyelination in the Chronic Phase of Severe Traumatic Brain Injury. Cells. 2023; 12(5):705. https://doi.org/10.3390/cells12050705
Chicago/Turabian StyleQiu, Xuecheng, Suning Ping, Michele Kyle, Lawrence Chin, and Li-Ru Zhao. 2023. "Stem Cell Factor and Granulocyte Colony-Stimulating Factor Promote Remyelination in the Chronic Phase of Severe Traumatic Brain Injury" Cells 12, no. 5: 705. https://doi.org/10.3390/cells12050705