
No Time to Die—How Islets Meet Their Demise in Transplantation



{kind=link}
Abstract
1. Introduction
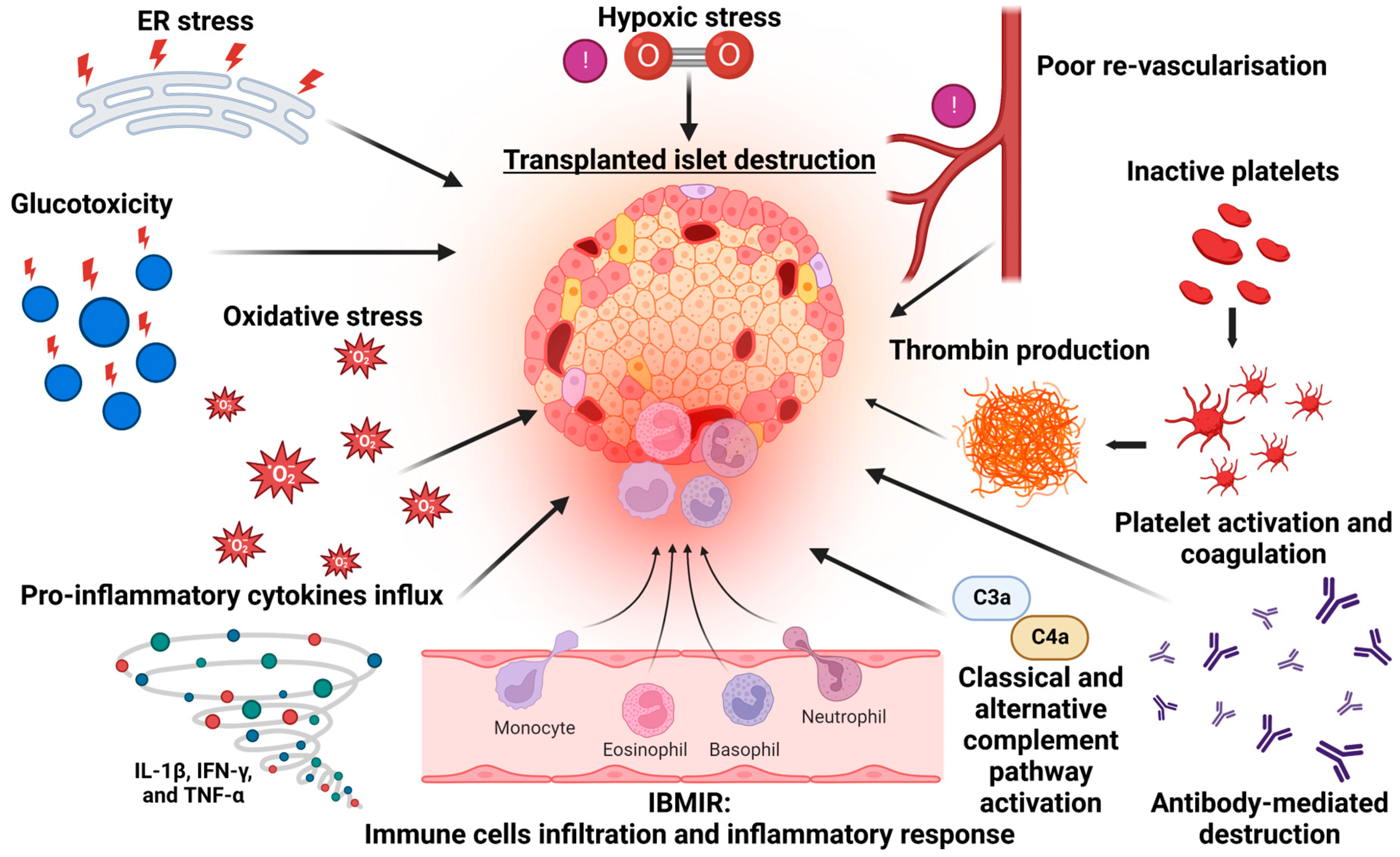
2. Mechanisms of Islet Cell Death in Islet Transplantation
3. Instant Blood Inflammatory Reaction (IBMIR) and Early Islet Demise
4. Alloimmunity and the Risk of Islet Transplant Failure
5. Non-Immunological Causes of Islet Death
6. The Importance of Islet–Endothelial Crosstalk
7. Future Directions in Islet Transplantation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martin, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- O’Connell, P.J.; Holmes-Walker, D.J.; Goodman, D.; Hawthorne, W.J.; Loudovaris, T.; Gunton, J.E.; Thomas, H.E.; Grey, S.T.; Drogemuller, C.J.; Ward, G.M.; et al. Multicenter Australian trial of islet transplantation: Improving accessibility and outcomes. Am. J. Transplant. 2013, 13, 1850–1858. [Google Scholar] [CrossRef]
- Gruessner, A.C. 2011 update on pancreas transplantation: Comprehensive trend analysis of 25,000 cases followed up over the course of twenty-four years at the International Pancreas Transplant Registry (IPTR). Rev. Diabet. Stud. 2011, 8, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Martín-Timón, I.; del Cañizo-Gómez, F.J. Mechanisms of hypoglycemia unawareness and implications in diabetic patients. World J. Diabetes 2015, 6, 912. [Google Scholar] [CrossRef] [PubMed]
- Hering, B.J.; Clarke, W.R.; Bridges, N.D.; Eggerman, T.L.; Alejandro, R.; Bellin, M.D.; Chaloner, K.; Czarniecki, C.W.; Goldstein, J.S.; Hunsicker, L.G. Phase 3 trial of transplantation of human islets in type 1 diabetes complicated by severe hypoglycemia. Diabetes Care 2016, 39, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.; Schopman, J.E.; Zammitt, N.N.; Frier, B.M. Prevalence of impaired awareness of hypoglycaemia in adults with Type 1 diabetes. Diabet. Med. 2008, 25, 501–504. [Google Scholar] [CrossRef]
- Chittineni, C.; Driver, B.E.; Halverson, M.; Cole, J.B.; Prekker, M.E.; Pandey, V.; Lai, T.; Harrington, J.; Zhao, S.; Klein, L.R. Incidence and causes of iatrogenic hypoglycemia in the emergency department. West. J. Emerg. Med. 2019, 20, 833. [Google Scholar] [CrossRef]
- Kalra, S.; Mukherjee, J.J.; Venkataraman, S.; Bantwal, G.; Shaikh, S.; Saboo, B.; Das, A.K.; Ramachandran, A. Hypoglycemia: The neglected complication. Indian J. Endocrinol. Metab. 2013, 17, 819. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef]
- Poggioli, R.; Faradji, R.N.; Ponte, G.; Betancourt, A.; Messinger, S.; Baidal, D.A.; Froud, T.; Ricordi, C.; Alejandro, R. Quality of life after islet transplantation. Am. J. Transplant. 2006, 6, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Wallner, K.; Shapiro, A.M.; Senior, P.A.; McCabe, C. Cost effectiveness and value of information analyses of islet cell transplantation in the management of ‘unstable’ type 1 diabetes mellitus. BMC Endocr Disord 2016, 16, 17. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Registry, C.I.T. Elevneth Allograft Report; CITR Coordinating Centre: Rockville, MD, USA, 2022. [Google Scholar]
- Eriksson, O.; Eich, T.; Sundin, A.; Tibell, A.; Tufveson, G.; Andersson, H.; Felldin, M.; Foss, A.; Kyllonen, L.; Langstrom, B.; et al. Positron emission tomography in clinical islet transplantation. Am. J. Transplant. 2009, 9, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Moberg, L.; Johansson, H.; Lukinius, A.; Berne, C.; Foss, A.; Kallen, R.; Ostraat, O.; Salmela, K.; Tibell, A.; Tufveson, G.; et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet 2002, 360, 2039–2045. [Google Scholar] [CrossRef]
- Bruni, A.; Bornstein, S.; Linkermann, A.; Shapiro, A.M.J. Regulated Cell Death Seen through the Lens of Islet Transplantation. Cell Transplant. 2018, 27, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Goto, M.; Dufrane, D.; Siegbahn, A.; Elgue, G.; Gianello, P.; Korsgren, O.; Nilsson, B. Low molecular weight dextran sulfate: A strong candidate drug to block IBMIR in clinical islet transplantation. Am. J. Transplant. 2006, 6, 305–312. [Google Scholar] [CrossRef]
- Verhoeff, K.; Marfil-Garza, B.A.; Sandha, G.; Cooper, D.; Dajani, K.; Bigam, D.L.; Anderson, B.; Kin, T.; Lam, A.; O’Gorman, D.; et al. Outcomes Following Extrahepatic and Intraportal Pancreatic Islet Transplantation: A Comparative Cohort Study. Transplantation 2022, 106, 2224–2231. [Google Scholar] [CrossRef]
- Gamble, A.; Pepper, A.R.; Bruni, A.; Shapiro, A.M.J. The journey of islet cell transplantation and future development. Islets 2018, 10, 80–94. [Google Scholar] [CrossRef]
- Hardstedt, M.; Lindblom, S.; Karlsson-Parra, A.; Nilsson, B.; Korsgren, O. Characterization of Innate Immunity in an Extended Whole Blood Model of Human Islet Allotransplantation. Cell Transplant. 2016, 25, 503–515. [Google Scholar] [CrossRef]
- Ishiyama, K.; Rawson, J.; Omori, K.; Mullen, Y. Liver natural killer cells play a role in the destruction of islets after intraportal transplantation. Transplantation 2011, 91, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y.; Ishiyama, K.; Ishida, N.; Tanaka, Y.; Ohdan, H. Memory-like Liver Natural Killer Cells are Responsible for Islet Destruction in Secondary Islet Transplantation. Sci. Rep. 2019, 9, 1022. [Google Scholar] [CrossRef] [PubMed]
- Kanak, M.A.; Takita, M.; Itoh, T.; SoRelle, J.A.; Murali, S.; Kunnathodi, F.; Shahbazov, R.; Lawrence, M.C.; Levy, M.F.; Naziruddin, B. Alleviation of instant blood-mediated inflammatory reaction in autologous conditions through treatment of human islets with NF-kappaB inhibitors. Transplantation 2014, 98, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, K.M.; Mysore, T.B.; Crikis, S.; Robson, S.C.; Nandurkar, H.; Cowan, P.J.; D’Apice, A.J. The transgenic expression of human CD39 on murine islets inhibits clotting of human blood. Transplantation 2006, 82, 428–432. [Google Scholar] [CrossRef]
- Tokodai, K.; Goto, M.; Inagaki, A.; Nakanishi, W.; Okada, N.; Okada, H.; Satomi, S. C5a-inhibitory peptide combined with gabexate mesilate prevents the instant blood-mediated inflammatory reaction in a rat model of islet transplantation. Transplant. Proc. 2010, 42, 2102–2103. [Google Scholar] [CrossRef]
- Goto, M.; Tjernberg, J.; Dufrane, D.; Elgue, G.; Brandhorst, D.; Ekdahl, K.N.; Brandhorst, H.; Wennberg, L.; Kurokawa, Y.; Satomi, S.; et al. Dissecting the instant blood-mediated inflammatory reaction in islet xenotransplantation. Xenotransplantation 2008, 15, 225–234. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Sumoski, W.; Rajotte, R.V.; Warnock, G.L. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J. Clin. Endocrinol. Metab. 1990, 71, 152–156. [Google Scholar] [CrossRef]
- Arnush, M.; Scarim, A.L.; Heitmeier, M.R.; Kelly, C.B.; Corbett, J.A. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J. Immunol. 1998, 160, 2684–2691. [Google Scholar] [CrossRef]
- Corbett, J.A.; McDaniel, M.L. Reversibility of interleukin-1 beta-induced islet destruction and dysfunction by the inhibition of nitric oxide synthase. Biochem. J. 1994, 299 Pt 3, 719–724. [Google Scholar] [CrossRef]
- Corbett, J.A.; Wang, J.L.; Hughes, J.H.; Wolf, B.A.; Sweetland, M.A.; Lancaster, J.R., Jr.; McDaniel, M.L. Nitric oxide and cyclic GMP formation induced by interleukin 1 beta in islets of Langerhans. Evidence for an effector role of nitric oxide in islet dysfunction. Biochem. J. 1992, 287 Pt 1, 229–235. [Google Scholar] [CrossRef]
- Johansson, U.; Olsson, A.; Gabrielsson, S.; Nilsson, B.; Korsgren, O. Inflammatory mediators expressed in human islets of Langerhans: Implications for islet transplantation. Biochem. Biophys. Res. Commun. 2003, 308, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Shim, W.; Oh, B.J.; Oh, S.H.; Yu, S.J.; Choi, J.M.; Park, H.J.; Park, J.B.; Kim, J.H. Anakinra Protects Against Serum Deprivation-Induced Inflammation and Functional Derangement in Islets Isolated From Nonhuman Primates. Am. J. Transplant. 2017, 17, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Montolio, M.; Biarnes, M.; Tellez, N.; Escoriza, J.; Soler, J.; Montanya, E. Interleukin-1beta and inducible form of nitric oxide synthase expression in early syngeneic islet transplantation. J. Endocrinol. 2007, 192, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, D.; Brandhorst, H.; Acreman, S.; Abraham, A.; Johnson, P.R.V. High Concentrations of Etanercept Reduce Human Islet Function and Integrity. J. Inflamm. Res. 2021, 14, 599–610. [Google Scholar] [CrossRef]
- Hering, B.J.; Kandaswamy, R.; Ansite, J.D.; Eckman, P.M.; Nakano, M.; Sawada, T.; Matsumoto, I.; Ihm, S.H.; Zhang, H.J.; Parkey, J.; et al. Single-donor, marginal-dose islet transplantation in patients with type 1 diabetes. JAMA 2005, 293, 830–835. [Google Scholar] [CrossRef]
- McCall, M.; Pawlick, R.; Kin, T.; Shapiro, A.M. Anakinra potentiates the protective effects of etanercept in transplantation of marginal mass human islets in immunodeficient mice. Am. J. Transplant. 2012, 12, 322–329. [Google Scholar] [CrossRef]
- Szempruch, K.R.; Banerjee, O.; McCall, R.C.; Desai, C.S. Use of anti-inflammatory agents in clinical islet cell transplants: A qualitative systematic analysis. Islets 2019, 11, 65–75. [Google Scholar] [CrossRef]
- Naziruddin, B.; Kanak, M.A.; Chang, C.A.; Takita, M.; Lawrence, M.C.; Dennison, A.R.; Onaca, N.; Levy, M.F. Improved outcomes of islet autotransplant after total pancreatectomy by combined blockade of IL-1beta and TNFalpha. Am. J. Transplant. 2018, 18, 2322–2329. [Google Scholar] [CrossRef]
- Takita, M.; Matsumoto, S.; Shimoda, M.; Chujo, D.; Itoh, T.; Sorelle, J.A.; Purcell, K.; Onaca, N.; Naziruddin, B.; Levy, M.F. Safety and tolerability of the T-cell depletion protocol coupled with anakinra and etanercept for clinical islet cell transplantation. Clin. Transplant. 2012, 26, E471–E484. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Batra, L.; Tariq Malik, M.; Tan, M.; Yolcu, E.S.; Shirwan, H. Immune checkpoint CD47 molecule engineered islets mitigate instant blood-mediated inflammatory reaction and show improved engraftment following intraportal transplantation. Am. J. Transplant. 2020, 20, 2703–2714. [Google Scholar] [CrossRef]
- Reffet, S.; Thivolet, C. Immunology of pancreatic islet transplantation. Diabetes Metab. 2006, 32, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Huurman, V.A.; Hilbrands, R.; Pinkse, G.G.; Gillard, P.; Duinkerken, G.; van de Linde, P.; van der Meer-Prins, P.M.; Versteeg-van der Voort Maarschalk, M.F.; Verbeeck, K.; Alizadeh, B.Z.; et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS ONE 2008, 3, e2435. [Google Scholar] [CrossRef]
- Regnell, S.E.; Lernmark, A. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Scirpoli, M.; Maffi, P.; Ghidoli, N.; De Taddeo, F.; Bertuzzi, F.; Piemonti, L.; Falcone, M.; Secchi, A.; Bonifacio, E. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J. Clin. Investig. 2008, 118, 1806–1814. [Google Scholar] [CrossRef]
- Pratt, J.R.; Basheer, S.A.; Sacks, S.H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 2002, 8, 582–587. [Google Scholar] [CrossRef]
- Brown, K.M.; Kondeatis, E.; Vaughan, R.W.; Kon, S.P.; Farmer, C.K.; Taylor, J.D.; He, X.; Johnston, A.; Horsfield, C.; Janssen, B.J.; et al. Influence of donor C3 allotype on late renal-transplantation outcome. N. Engl. J. Med. 2006, 354, 2014–2023. [Google Scholar] [CrossRef]
- Lawrence, C.; Willicombe, M.; Brookes, P.A.; Santos-Nunez, E.; Bajaj, R.; Cook, T.; Roufosse, C.; Taube, D.; Warrens, A.N. Preformed complement-activating low-level donor-specific antibody predicts early antibody-mediated rejection in renal allografts. Transplantation 2013, 95, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Toquet, C.; Rouvier, P.; Beuscart, T.; Bories, M.C.; Varnous, S.; Guillemain, R.; Pattier, S.; Suberbielle, C.; Leprince, P.; et al. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. Am. J. Transplant. 2016, 16, 111–120. [Google Scholar] [CrossRef]
- Roux, A.; Bendib Le Lan, I.; Holifanjaniaina, S.; Thomas, K.A.; Picard, C.; Grenet, D.; De Miranda, S.; Douvry, B.; Beaumont-Azuar, L.; Sage, E.; et al. Characteristics of Donor-Specific Antibodies Associated With Antibody-Mediated Rejection in Lung Transplantation. Front. Med. 2017, 4, 155. [Google Scholar] [CrossRef]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Jackson, A.M.; et al. Evidence for Induced Expression of HLA Class II on Human Islets: Possible Mechanism for HLA Sensitization in Transplant Recipients. Transplantation 2009, 87, 500–506. [Google Scholar] [CrossRef]
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895. [Google Scholar] [CrossRef]
- Dai, H.; Zheng, Y.; Thomson, A.W.; Rogers, N.M. Transplant Tolerance Induction: Insights From the Liver. Front. Immunol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Piemonti, L.; Everly, M.J.; Maffi, P.; Scavini, M.; Poli, F.; Nano, R.; Cardillo, M.; Melzi, R.; Mercalli, A.; Sordi, V.; et al. Alloantibody and autoantibody monitoring predicts islet transplantation outcome in human type 1 diabetes. Diabetes 2013, 62, 1656–1664. [Google Scholar] [CrossRef]
- Brooks, A.M.; Carter, V.; Liew, A.; Marshall, H.; Aldibbiat, A.; Sheerin, N.S.; Manas, D.M.; White, S.A.; Shaw, J.A. De Novo Donor-Specific HLA Antibodies Are Associated With Rapid Loss of Graft Function Following Islet Transplantation in Type 1 Diabetes. Am. J. Transplant. 2015, 15, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Naziruddin, B.; Wease, S.; Stablein, D.; Barton, F.B.; Berney, T.; Rickels, M.R.; Alejandro, R. HLA class I sensitization in islet transplant recipients: Report from the Collaborative Islet Transplant Registry. Cell Transplant. 2012, 21, 901–908. [Google Scholar] [CrossRef]
- Pouliquen, E.; Baltzinger, P.; Lemle, A.; Chen, C.C.; Parissiadis, A.; Borot, S.; Frimat, L.; Girerd, S.; Berney, T.; Lablanche, S.; et al. Anti-Donor HLA Antibody Response After Pancreatic Islet Grafting: Characteristics, Risk Factors, and Impact on Graft Function. Am. J. Transplant. 2017, 17, 462–473. [Google Scholar] [CrossRef]
- Chen, C.C.; Pouliquen, E.; Broisat, A.; Andreata, F.; Racape, M.; Bruneval, P.; Kessler, L.; Ahmadi, M.; Bacot, S.; Saison-Delaplace, C.; et al. Endothelial chimerism and vascular sequestration protect pancreatic islet grafts from antibody-mediated rejection. J. Clin. Investig. 2018, 128, 219–232. [Google Scholar] [CrossRef]
- Heit, J.J.; Apelqvist, A.A.; Gu, X.; Winslow, M.M.; Neilson, J.R.; Crabtree, G.R.; Kim, S.K. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006, 443, 345–349. [Google Scholar] [CrossRef]
- Xu, C.; Niu, Y.J.; Liu, X.J.; Teng, Y.Q.; Li, C.F.; Wang, H.Y.; Yin, J.P.; Wang, L.T.; Shen, Z.Y. Tacrolimus reversibly reduces insulin secretion, induces insulin resistance, and causes islet cell damage in rats. Int. J. Clin. Pharmacol. Ther. 2014, 52, 620–627. [Google Scholar] [CrossRef]
- Dai, C.; Walker, J.T.; Shostak, A.; Padgett, A.; Spears, E.; Wisniewski, S.; Poffenberger, G.; Aramandla, R.; Dean, E.D.; Prasad, N.; et al. Tacrolimus- and sirolimus-induced human beta cell dysfunction is reversible and preventable. JCI Insight 2020, 5, e130770. [Google Scholar] [CrossRef]
- Johnson, J.D.; Ao, Z.; Ao, P.; Li, H.; Dai, L.J.; He, Z.; Tee, M.; Potter, K.J.; Klimek, A.M.; Meloche, R.M.; et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009, 18, 833–845. [Google Scholar] [CrossRef]
- Jhala, U.S.; Canettieri, G.; Screaton, R.A.; Kulkarni, R.N.; Krajewski, S.; Reed, J.; Walker, J.; Lin, X.; White, M.; Montminy, M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003, 17, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Soleimanpour, S.A.; Crutchlow, M.F.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Rankin, M.M.; Liu, C.; De Leon, D.D.; Naji, A.; Kushner, J.A.; et al. Calcineurin signaling regulates human islet beta-cell survival. J. Biol. Chem. 2010, 285, 40050–40059. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. Cyclosporine A and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: Increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J. Clin. Endocrinol. Metab. 2014, 99, E1885–E1894. [Google Scholar] [CrossRef]
- Lombardi, A.; Trimarco, B.; Iaccarino, G.; Santulli, G. Impaired mitochondrial calcium uptake caused by tacrolimus underlies beta-cell failure. Cell Commun. Signal. 2017, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Hribal, M.L.; Lin, H.V.; Bennett, W.R.; Ward, A.; Accili, D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J. Clin. Investig. 2006, 116, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Trinanes, J.; Rodriguez-Rodriguez, A.E.; Brito-Casillas, Y.; Wagner, A.; De Vries, A.P.J.; Cuesto, G.; Acebes, A.; Salido, E.; Torres, A.; Porrini, E. Deciphering Tacrolimus-Induced Toxicity in Pancreatic beta Cells. Am. J. Transplant. 2017, 17, 2829–2840. [Google Scholar] [CrossRef]
- Ekberg, H.; Tedesco-Silva, H.; Demirbas, A.; Vitko, S.; Nashan, B.; Gurkan, A.; Margreiter, R.; Hugo, C.; Grinyo, J.M.; Frei, U.; et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N. Engl. J. Med. 2007, 357, 2562–2575. [Google Scholar] [CrossRef]
- Vincenti, F.; Friman, S.; Scheuermann, E.; Rostaing, L.; Jenssen, T.; Campistol, J.M.; Uchida, K.; Pescovitz, M.D.; Marchetti, P.; Tuncer, M.; et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am. J. Transplant. 2007, 7, 1506–1514. [Google Scholar] [CrossRef]
- Johnston, O.; Rose, C.L.; Webster, A.C.; Gill, J.S. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J. Am. Soc. Nephrol. 2008, 19, 1411–1418. [Google Scholar] [CrossRef]
- Fraenkel, M.; Ketzinel-Gilad, M.; Ariav, Y.; Pappo, O.; Karaca, M.; Castel, J.; Berthault, M.F.; Magnan, C.; Cerasi, E.; Kaiser, N.; et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008, 57, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, M.S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract 2007, 77 (Suppl. 1), S49–S57. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Blattler, S.M.; Cunningham, J.T.; Verdeguer, F.; Chim, H.; Haas, W.; Liu, H.; Romanino, K.; Ruegg, M.A.; Gygi, S.P.; Shi, Y.; et al. Yin Yang 1 deficiency in skeletal muscle protects against rapamycin-induced diabetic-like symptoms through activation of insulin/IGF signaling. Cell Metab. 2012, 15, 505–517. [Google Scholar] [CrossRef]
- Bell, E.; Cao, X.; Moibi, J.A.; Greene, S.R.; Young, R.; Trucco, M.; Gao, Z.; Matschinsky, F.M.; Deng, S.; Markman, J.F.; et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes 2003, 52, 2731–2739. [Google Scholar] [CrossRef]
- Bussiere, C.T.; Lakey, J.R.; Shapiro, A.M.; Korbutt, G.S. The impact of the mTOR inhibitor sirolimus on the proliferation and function of pancreatic islets and ductal cells. Diabetologia 2006, 49, 2341–2349. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Toso, C.; Isse, K.; Demetris, A.J.; Dinyari, P.; Koh, A.; Imes, S.; Kin, T.; Emamaullee, J.; Senior, P.; Shapiro, A.M. Histologic graft assessment after clinical islet transplantation. Transplantation 2009, 88, 1286–1293. [Google Scholar] [CrossRef]
- Duijnhoven, E.M.V.; Boots, J.M.M.; Christiaans, M.H.L.; Wolffenbuttel, B.H.R.; Hooff, J.P.V. Influence of tacrolimus on glucose metabolism before and after renal transplantation: A prospective study. J. Am. Soc. Nephrol. 2001, 12, 583–588. [Google Scholar] [CrossRef]
- Westermark, G.T.; Westermark, P.; Berne, C.; Korsgren, O.; Nordic Network for Clinical Islet, T. Widespread amyloid deposition in transplanted human pancreatic islets. N. Engl. J. Med. 2008, 359, 977–979. [Google Scholar] [CrossRef]
- Udayasankar, J.; Kodama, K.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Subramanian, S.L.; Tong, J.; Faulenbach, M.V.; Vidal, J.; Kahn, S.E. Amyloid formation results in recurrence of hyperglycaemia following transplantation of human IAPP transgenic mouse islets. Diabetologia 2009, 52, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.J.; Werner, I.; Denroche, H.C.; Montane, J.; Plesner, A.; Chen, Y.; Lei, D.; Soukhatcheva, G.; Warnock, G.L.; Oberholzer, J.; et al. Amyloid formation in human islets is enhanced by heparin and inhibited by heparinase. Am. J. Transplant. 2015, 15, 1519–1530. [Google Scholar] [CrossRef]
- Henderson, J.R.; Moss, M.C. A morphometric study of the endocrine and exocrine capillaries of the pancreas. Q. J. Exp. Physiol. 1985, 70, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Shostak, A.; Shiota, M.; Wiebe, P.O.; Poffenberger, G.; Kantz, J.; Chen, Z.; Carr, C.; Jerome, W.G.; Chen, J.; et al. Pancreatic islet production of vascular endothelial growth factor—A is essential for islet vascularization, revascularization, and function. Diabetes 2006, 55, 2974–2985. [Google Scholar] [CrossRef]
- Su, D.; Zhang, N.; He, J.; Qu, S.; Slusher, S.; Bottino, R.; Bertera, S.; Bromberg, J.; Dong, H.H. Angiopoietin-1 production in islets improves islet engraftment and protects islets from cytokine-induced apoptosis. Diabetes 2007, 56, 2274–2283. [Google Scholar] [CrossRef]
- Dorrell, C.; Schug, J.; Lin, C.F.; Canaday, P.S.; Fox, A.J.; Smirnova, O.; Bonnah, R.; Streeter, P.R.; Stoeckert, C.J., Jr.; Kaestner, K.H.; et al. Transcriptomes of the major human pancreatic cell types. Diabetologia 2011, 54, 2832–2844. [Google Scholar] [CrossRef]
- Konstantinova, I.; Nikolova, G.; Ohara-Imaizumi, M.; Meda, P.; Kucera, T.; Zarbalis, K.; Wurst, W.; Nagamatsu, S.; Lammert, E. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 2007, 129, 359–370. [Google Scholar] [CrossRef]
- Kuboki, K.; Jiang, Z.Y.; Takahara, N.; Ha, S.W.; Igarashi, M.; Yamauchi, T.; Feener, E.P.; Herbert, T.P.; Rhodes, C.J.; King, G.L. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: A specific vascular action of insulin. Circulation 2000, 101, 676–681. [Google Scholar] [CrossRef]
- Wang, R.N.; Rosenberg, L. Maintenance of beta-cell function and survival following islet isolation requires re-establishment of the islet-matrix relationship. J. Endocrinol. 1999, 163, 181–190. [Google Scholar] [CrossRef]
- Thomas, F.; Wu, J.; Contreras, J.L.; Smyth, C.; Bilbao, G.; He, J.; Thomas, J. A tripartite anoikis-like mechanism causes early isolated islet apoptosis. Surgery 2001, 130, 333–338. [Google Scholar] [CrossRef]
- Irving-Rodgers, H.F.; Choong, F.J.; Hummitzsch, K.; Parish, C.R.; Rodgers, R.J.; Simeonovic, C.J. Pancreatic islet basement membrane loss and remodeling after mouse islet isolation and transplantation: Impact for allograft rejection. Cell Transplant. 2014, 23, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, R.; Zuellig, R.A.; Kugelmeier, P.; Baenninger, P.B.; Moritz, W.; Perren, A.; Clavien, P.A.; Weber, M.; Spinas, G.A. Superiority of small islets in human islet transplantation. Diabetes 2007, 56, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Suszynski, T.M.; Wilhelm, J.J.; Radosevich, D.M.; Balamurugan, A.N.; Sutherland, D.E.; Beilman, G.J.; Dunn, T.B.; Chinnakotla, S.; Pruett, T.L.; Vickers, S.M.; et al. Islet size index as a predictor of outcomes in clinical islet autotransplantation. Transplantation 2014, 97, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Olerud, J.; Pettersson, U.; Carlsson, P.-O. Increased numbers of low-oxygenated pancreatic islets after intraportal islet transplantation. Diabetes 2011, 60, 2350–2353. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Cook, C.; Wang, C.H.; Medrano, L.; Lin, H.; Kandeel, F.; Tai, Y.C.; Mullen, Y. Oxygen environment and islet size are the primary limiting factors of isolated pancreatic islet survival. PLoS ONE 2017, 12, e0183780. [Google Scholar] [CrossRef]
- Addison, P.; Fatakhova, K.; Rodriguez Rilo, H.L. Considerations for an Alternative Site of Islet Cell Transplantation. J. Diabetes Sci. Technol. 2020, 14, 338–344. [Google Scholar] [CrossRef]
- Kanak, M.A.; Takita, M.; Kunnathodi, F.; Lawrence, M.C.; Levy, M.F.; Naziruddin, B. Inflammatory response in islet transplantation. Int. J. Endocrinol. 2014, 2014, 451035. [Google Scholar] [CrossRef]
- Miao, G.; Ostrowski, R.P.; Mace, J.; Hough, J.; Hopper, A.; Peverini, R.; Chinnock, R.; Zhang, J.; Hathout, E. Dynamic production of hypoxia-inducible factor-1alpha in early transplanted islets. Am. J. Transplant. 2006, 6, 2636–2643. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef]
- Stokes, R.A.; Cheng, K.; Deters, N.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Stolp, J.; Grey, S.; Loudovaris, T.; Kay, T.W.; et al. Hypoxia-inducible factor-1alpha (HIF-1alpha) potentiates beta-cell survival after islet transplantation of human and mouse islets. Cell Transplant. 2013, 22, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Dionne, K.E.; Colton, C.K.; Yarmush, M.L. Effect of hypoxia on insulin secretion by isolated rat and canine islets of Langerhans. Diabetes 1993, 42, 12–21. [Google Scholar] [CrossRef]
- Zehetner, J.; Danzer, C.; Collins, S.; Eckhardt, K.; Gerber, P.A.; Ballschmieter, P.; Galvanovskis, J.; Shimomura, K.; Ashcroft, F.M.; Thorens, B.; et al. PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev. 2008, 22, 3135–3146. [Google Scholar] [CrossRef] [PubMed]
- Cantley, J.; Walters, S.N.; Jung, M.H.; Weinberg, A.; Cowley, M.J.; Whitworth, T.P.; Kaplan, W.; Hawthorne, W.J.; O’Connell, P.J.; Weir, G.; et al. A preexistent hypoxic gene signature predicts impaired islet graft function and glucose homeostasis. Cell Transplant. 2013, 22, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, X.; Wang, X.; Ma, Z.; Gupta Sunkari, V.; Botusan, I.; Takeda, T.; Bjorklund, A.; Inoue, M.; Catrina, S.B.; et al. Acute hypoxia induces apoptosis of pancreatic beta-cell by activation of the unfolded protein response and upregulation of CHOP. Cell Death Dis. 2012, 3, e322. [Google Scholar] [CrossRef]
- Kataoka, H.U.; Noguchi, H. ER Stress and beta-Cell Pathogenesis of Type 1 and Type 2 Diabetes and Islet Transplantation. Cell Med. 2013, 5, 53–57. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef]
- Walters, S.N.; Luzuriaga, J.; Chan, J.Y.; Grey, S.T.; Laybutt, D.R. Influence of chronic hyperglycemia on the loss of the unfolded protein response in transplanted islets. J. Mol. Endocrinol. 2013, 51, 225–232. [Google Scholar] [CrossRef]
- Bensellam, M.; Maxwell, E.L.; Chan, J.Y.; Luzuriaga, J.; West, P.K.; Jonas, J.C.; Gunton, J.E.; Laybutt, D.R. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia 2016, 59, 1492–1502. [Google Scholar] [CrossRef]
- Schaschkow, A.; Sigrist, S.; Mura, C.; Dissaux, C.; Bouzakri, K.; Lejay, A.; Bruant-Rodier, C.; Pinget, M.; Maillard, E. Extra-Hepatic Islet Transplantation: Validation of the h-Omental Matrix Islet filliNG (hOMING) Technique on a Rodent Model Using an Alginate Carrier. Cell Transplant. 2018, 27, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Buitinga, M.; Truckenmuller, R.; Engelse, M.A.; Moroni, L.; Ten Hoopen, H.W.; van Blitterswijk, C.A.; de Koning, E.J.; van Apeldoorn, A.A.; Karperien, M. Microwell scaffolds for the extrahepatic transplantation of islets of Langerhans. PLoS ONE 2013, 8, e64772. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, E.; Brady, A.C.; Fraker, C.A.; Molano, R.D.; Sukert, S.; Berman, D.M.; Kenyon, N.S.; Pileggi, A.; Ricordi, C.; Stabler, C.L. Macroporous three-dimensional PDMS scaffolds for extrahepatic islet transplantation. Cell Transplant. 2013, 22, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Cayabyab, F.; Nih, L.R.; Yoshihara, E. Advances in Pancreatic Islet Transplantation Sites for the Treatment of Diabetes. Front. Endocrinol. 2021, 12, 732431. [Google Scholar] [CrossRef]
- Hirabaru, M.; Kuroki, T.; Adachi, T.; Kitasato, A.; Ono, S.; Tanaka, T.; Matsushima, H.; Sakai, Y.; Soyama, A.; Hidaka, M.; et al. A Method for Performing Islet Transplantation Using Tissue-Engineered Sheets of Islets and Mesenchymal Stem Cells. Tissue Eng. Part C Methods 2015, 21, 1205–1215. [Google Scholar] [CrossRef]
- Won, K.C.; Moon, J.S.; Eun, M.J.; Yoon, J.S.; Chun, K.A.; Cho, I.H.; Kim, Y.W.; Lee, H.W. A protective role for heme oxygenase-1 in INS-1 cells and rat islets that are exposed to high glucose conditions. J. Korean Med. Sci. 2006, 21, 418–424. [Google Scholar] [CrossRef]
- Plesner, A.; Soukhatcheva, G.; Korneluk, R.G.; Verchere, C.B. XIAP inhibition of beta-cell apoptosis reduces the number of islets required to restore euglycemia in a syngeneic islet transplantation model. Islets 2010, 2, 18–23. [Google Scholar] [CrossRef]
- Pileggi, A.; Molano, R.D.; Berney, T.; Cattan, P.; Vizzardelli, C.; Oliver, R.; Fraker, C.; Ricordi, C.; Pastori, R.L.; Bach, F.H.; et al. Heme oxygenase-1 induction in islet cells results in protection from apoptosis and improved in vivo function after transplantation. Diabetes 2001, 50, 1983–1991. [Google Scholar] [CrossRef]
- Yamashita, M.; Adachi, T.; Adachi, T.; Ono, S.; Matsumura, N.; Maekawa, K.; Sakai, Y.; Hidaka, M.; Kanetaka, K.; Kuroki, T.; et al. Subcutaneous transplantation of engineered islet/adipose-derived mesenchymal stem cell sheets in diabetic pigs with total pancreatectomy. Regen. Ther. 2021, 16, 42–52. [Google Scholar] [CrossRef]
- Matsushima, H.; Kuroki, T.; Adachi, T.; Kitasato, A.; Ono, S.; Tanaka, T.; Hirabaru, M.; Kuroshima, N.; Hirayama, T.; Sakai, Y.; et al. Human Fibroblast Sheet Promotes Human Pancreatic Islet Survival and Function In Vitro. Cell Transplant. 2016, 25, 1525–1537. [Google Scholar] [CrossRef]
- Narayanan, S.; Loganathan, G.; Dhanasekaran, M.; Tucker, W.; Patel, A.; Subhashree, V.; Mokshagundam, S.; Hughes, M.G.; Williams, S.K.; Balamurugan, A.N. Intra-islet endothelial cell and beta-cell crosstalk: Implication for islet cell transplantation. World J. Transplant. 2017, 7, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, P.; Antonini, S.; Spiga, S.; Mazzanti, B.; Curcio, M.; Mulas, G.; Diana, M.; Marzola, P.; Mosca, F.; Longoni, B. Co-transplantation of endothelial progenitor cells and pancreatic islets to induce long-lasting normoglycemia in streptozotocin-treated diabetic rats. PLoS ONE 2014, 9, e94783. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.J.; Oh, S.H.; Jin, S.M.; Suh, S.; Bae, J.C.; Park, C.G.; Lee, M.S.; Lee, M.K.; Kim, J.H.; Kim, K.W. Co-transplantation of bone marrow-derived endothelial progenitor cells improves revascularization and organization in islet grafts. Am. J. Transplant. 2013, 13, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Park, H.S.; Jo, A.; Hong, S.H.; Lee, H.N.; Lee, Y.Y.; Park, J.S.; Jung, H.S.; Chung, S.S.; Park, K.S. Endothelial progenitor cell cotransplantation enhances islet engraftment by rapid revascularization. Diabetes 2012, 61, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Nalbach, L.; Roma, L.P.; Schmitt, B.M.; Becker, V.; Korbel, C.; Wrublewsky, S.; Pack, M.; Spater, T.; Metzger, W.; Menger, M.M.; et al. Improvement of islet transplantation by the fusion of islet cells with functional blood vessels. EMBO Mol. Med. 2021, 13, e12616. [Google Scholar] [CrossRef]
- Wrublewsky, S.; Weinzierl, A.; Hornung, I.; Prates-Roma, L.; Menger, M.D.; Laschke, M.W.; Ampofo, E. Co-transplantation of pancreatic islets and microvascular fragments effectively restores normoglycemia in diabetic mice. NPJ Regen. Med. 2022, 7, 67. [Google Scholar] [CrossRef]
- McGuigan, A.P.; Sefton, M.V. Vascularized organoid engineered by modular assembly enables blood perfusion. Proc. Natl. Acad. Sci. USA 2006, 103, 11461–11466. [Google Scholar] [CrossRef]
- Vlahos, A.E.; Cober, N.; Sefton, M.V. Modular tissue engineering for the vascularization of subcutaneously transplanted pancreatic islets. Proc. Natl. Acad. Sci. USA 2017, 114, 9337–9342. [Google Scholar] [CrossRef]
- Zhang, Q.; Gonelle-Gispert, C.; Li, Y.; Geng, Z.; Gerber-Lemaire, S.; Wang, Y.; Buhler, L. Islet Encapsulation: New Developments for the Treatment of Type 1 Diabetes. Front. Immunol. 2022, 13, 869984. [Google Scholar] [CrossRef]
- Jacobs-Tulleneers-Thevissen, D.; Chintinne, M.; Ling, Z.; Gillard, P.; Schoonjans, L.; Delvaux, G.; Strand, B.L.; Gorus, F.; Keymeulen, B.; Pipeleers, D.; et al. Sustained function of alginate-encapsulated human islet cell implants in the peritoneal cavity of mice leading to a pilot study in a type 1 diabetic patient. Diabetologia 2013, 56, 1605–1614. [Google Scholar] [CrossRef]
- Basta, G.; Montanucci, P.; Luca, G.; Boselli, C.; Noya, G.; Barbaro, B.; Qi, M.; Kinzer, K.P.; Oberholzer, J.; Calafiore, R. Long-term metabolic and immunological follow-up of nonimmunosuppressed patients with type 1 diabetes treated with microencapsulated islet allografts: Four cases. Diabetes Care 2011, 34, 2406–2409. [Google Scholar] [CrossRef]
- Bochenek, M.A.; Veiseh, O.; Vegas, A.J.; McGarrigle, J.J.; Qi, M.; Marchese, E.; Omami, M.; Doloff, J.C.; Mendoza-Elias, J.; Nourmohammadzadeh, M.; et al. Alginate encapsulation as long-term immune protection of allogeneic pancreatic islet cells transplanted into the omental bursa of macaques. Nat. Biomed. Eng. 2018, 2, 810–821. [Google Scholar] [CrossRef]
- Leung, A.; Lawrie, G.; Nielsen, L.K.; Trau, M. Synthesis and characterization of alginate/poly-L-ornithine/alginate microcapsules for local immunosuppression. J. Microencapsul. 2008, 25, 387–398. [Google Scholar] [CrossRef]
- Li, J.; Thomson, A.W.; Rogers, N.M. Myeloid and Mesenchymal Stem Cell Therapies for Solid Organ Transplant Tolerance. Transplantation 2021, 105, e303–e321. [Google Scholar] [CrossRef]
- Wang, H.; Strange, C.; Nietert, P.J.; Wang, J.; Turnbull, T.L.; Cloud, C.; Owczarski, S.; Shuford, B.; Duke, T.; Gilkeson, G.; et al. Autologous Mesenchymal Stem Cell and Islet Cotransplantation: Safety and Efficacy. Stem Cells Transl. Med. 2018, 7, 11–19. [Google Scholar] [CrossRef]
- Reid, L.; Baxter, F.; Forbes, S. Effects of islet transplantation on microvascular and macrovascular complications in type 1 diabetes. Diabet. Med. 2021, 38, e14570. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kale, A.; Rogers, N.M. No Time to Die—How Islets Meet Their Demise in Transplantation. Cells 2023, 12, 796. https://doi.org/10.3390/cells12050796
Kale A, Rogers NM. No Time to Die—How Islets Meet Their Demise in Transplantation. Cells. 2023; 12(5):796. https://doi.org/10.3390/cells12050796
Chicago/Turabian StyleKale, Atharva, and Natasha M. Rogers. 2023. "No Time to Die—How Islets Meet Their Demise in Transplantation" Cells 12, no. 5: 796. https://doi.org/10.3390/cells12050796
APA StyleKale, A., & Rogers, N. M. (2023). No Time to Die—How Islets Meet Their Demise in Transplantation. Cells, 12(5), 796. https://doi.org/10.3390/cells12050796