
Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction: The Cellular Response to Hypoxia and the Role of HIFs
2. The HIF-Dependent Transcriptional Response
3. HIF-1-Interacting Co-Regulators

{kind=link}
{kind=link}
{kind=link}
| HIFα Isoform | HIF-1α Domain (Residues) Involved | Co-Regulators | Effectors | Ref. | Cell Types |
|---|---|---|---|---|---|
| Acetyl transferases | |||||
| HIF-1α | N-TAD (532–585) C-TAD (776–826) | p300/CBP (+) | [25,26,27,28] | HEK293, HeLa, Hep3B, HCT166 | |
| HIF-1α/2α | Asn-803/847 | FIH-1 (−) | [29,30,31] | HEK293, Hep3B | |
| HIF-1α | Lys-674 | SIRT1 (−) PCAF (+) | [32] | HEK293, Hep3B, HT1080 | |
| HIF-1α | ND | SRC-1 (+) Ref-1 (+) | [33] | HEK293, COS7 | |
| HIF-1α | ND | #MUC1 (+) | [34] | Pancreatic cancer | |
| HIF-1α | C-TAD | #PKA (+) | [35] | HeLa, cardiomyocytes | |
| HIF-1α | N-term. (1–400) | FABP5 (+) | [36] | HEK293, HepG2 | |
| HIF-1α | C-TAD | #CITED2 (−) | [37,38] | Hep3B | |
| HIF-1α | ODD/N-TAD (429–608) | FHL2 (−) | [39] | HEK293, Hep3B | |
| HIF-1α/2α | C-TAD | FGFR2 (−) | [40] | DU145, PC3 | |
| HIF-1α | C-TAD | EAF2 (−) | [41] | HEK293, ccRCC | |
| HIF-1α | Reptin (−) | #G9a (+) | [42] | MCF7 | |
| HIF-1α | ND | Pontin (+) | #G9a (+) GLP (+) | [43] | MCF7 |
| HIF-1α | TIP60 (+) | [44] | HCT116 | ||
| Methylation/demethylation enzymes | |||||
| HIF-1α | C-term. (575–826) | # JMJD2C (+) | [45] | HeLa, MDA-MB-435 | |
| HIF-1α | bHLH (17–70) | # JMJD1A (+) | [46,47] | HUVEC, UBC | |
| HIF-1α/2α | PAS-B (175–305) | #TET1 (+) | [48,49] | HEK293, H1299, FADU | |
| HIF-1α | ND | SET9 (+) | [50] | HEK293, Hep3B, U2OS | |
| HIF-1α/2α | N-term. (1–396) | SET1B (+) | [51] | HeLa, A549 | |
| Other epigenetic enzymes & Epigenetic readers | |||||
| HIF-1α/2α | ND | # PADI4 (+) | [52] | Breast cancer, Hepatoma | |
| HIF-1α/2α | ND | # ZMYND8 (+) | p300 (+) | [53] | Breast cancer |
| Transcriptional machinery | |||||
| HIF-1α | C-TAD | CDK8- Mediator (+) | AFF4 (+) CDK9 (+) | [54] | HCT116 |
| HIF-1α/2α | C-term. (531–826) | TRIM28 (+) | CDK9 (+) | [55] | Breast cancer |
| Chromatin remodeling factors | |||||
| HIF-1α/2α | ND | BRG1 (+) | [56] | HEK293, Hep3B | |
| HIF-1α/2α | C-term. (531–826) | CHD4 (+) | p300 (+) | [57] | Breast cancer |
| HIF-1α | ETD (616–658) Ser641/643 | # NPM1 (+) | ERK1/2 (+) | [58] | HeLa, Huh7 |
| Other proteins | |||||
| HIF-1α/2α | C-term. (531–826) | #PKM2 (+) | PHD3 (+) | [59] | HeLa, Hep3B, RCC4 |
| JMJD5 (+) | [60] | HeLa, MCF7 | |||
| Digoxin (−) | [61] | Macrophages | |||
| [62,63] | Macrophages | ||||
| HIF-1α/2α | C-term./ID (604–726) | FBP1 (−) | [64] | HEK293, ccRCC, HK-2, A549 | |
| HIF-1α/2α | ND | PARP1 (+) | [65] | K562, MLF | |
| HIF-1α | N-term. (1–390) | Filamin A (+) | [66] | Melanoma, HeLa NIH 3T3, COS1, HEK293, U2OS | |
3.1. CBP/p300
3.2. Protein Effectors Regulating the Interaction between HIF-1α and CBP/p300
3.2.1. Post-Translational Modifications Affecting the HIF-1α-CBP/p300 Interaction
3.2.2. Positive Protein Effectors of the HIF-1α-CBP/p300 Interaction
3.2.3. Negative Protein Effectors of the HIF-1α-CBP/p300 Interaction
3.3. The TIP60 Complex
3.4. Methylation & Demethylation Enzymes
3.5. Other Epigenetic Enzymes: PADI4
3.6. Epigenetic Readers: ZMYND8 & BRD4
3.7. Components of the Transcriptional Machinery
3.7.1. CDK8-Mediator
3.7.2. TRIM28/DNA-PK
3.8. Chromatin Remodeling Factors
3.8.1. BRG1
3.8.2. CHD4
3.8.3. NPM1
3.9. Other Proteins
3.9.1. PKM2
3.9.2. FBP1
3.9.3. PARP1
3.9.4. Filamin A
4. Defining a Core of HIF-1α-Associated HIF-1 Co-Activators
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Wu, C.-Y.; Yang, H.-Y. Discoveries of how cells sense oxygen win the 2019 Nobel Prize in Physiology or medicine. Biomed. J. 2020, 43, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. The Genomics and Genetics of Oxygen Homeostasis. Annu. Rev. Genom. Hum. Genet. 2020, 21, 183–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, E.S. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [Green Version]
- Beck, I.; Ramirez, S.; Weinmann, R.; Caro, J. Enhancer element at the 3’-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J. Biol. Chem. 1991, 266, 15563–15566. [Google Scholar] [CrossRef]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant Regulation of Gene Expression by Hypoxia and 2-Oxoglutarate-dependent Dioxygenase Inhibition. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef] [Green Version]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar] [CrossRef] [Green Version]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide Association of Hypoxia-inducible Factor (HIF)-1α and HIF-2α DNA Binding with Expression Profiling of Hypoxia-inducible Transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Kung, A.L. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009, 10, R113. [Google Scholar] [CrossRef]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; Del Peso, L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010, 38, 2332–2345. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Schödel, J.; Oikonomopoulos, S.; Camps, C.; Grampp, S.; Harris, A.; Ratcliffe, P.; Ragoussis, J.; Mole, D.R. Extensive regulation of the non-coding transcriptome by hypoxia: Role of HIF in releasing paused RNA pol2. EMBO Rep. 2013, 15, 70–76. [Google Scholar] [CrossRef]
- Chan, M.C.; Ilott, N.E.; Schödel, J.; Sims, D.; Tumber, A.; Lippl, K.; Mole, D.R.; Pugh, C.W.; Ratcliffe, P.J.; Ponting, C.P.; et al. Tuning the Transcriptional Response to Hypoxia by Inhibiting Hypoxia-inducible Factor (HIF) Prolyl and Asparaginyl Hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. [Google Scholar] [CrossRef] [Green Version]
- Smythies, A.J.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, E.M.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA -binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2018, 20, e46401. [Google Scholar] [CrossRef]
- Ye, I.C.; Fertig, E.J.; DiGiacomo, J.W.; Considine, M.; Godet, I.; Gilkes, D.M. Molecular Portrait of Hypoxia in Breast Cancer: A Prognostic Signature and Novel HIF-Regulated Genes. Mol. Cancer Res. 2018, 16, 1889–1901. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial Cell–Specific regulation of Hypoxia-Inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Kindrick, J.; Mole, D. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. Int. J. Mol. Sci. 2020, 21, 8320. [Google Scholar] [CrossRef]
- Batie, M.; Frost, J.; Shakir, D.; Rocha, S. Regulation of chromatin accessibility by hypoxia and HIF. Biochem. J. 2022, 479, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Vojnovic, N.; Lee, K.-L.; Yang, H.; Gradin, K.; Poellinger, L. HIF-dependent and reversible nucleosome disassembly in hypoxia-inducible gene promoters. Exp. Cell Res. 2018, 366, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. A compendium of proteins that interact with HIF-1α. Exp. Cell Res. 2017, 356, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Hu, C.-J. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell Signal. 2013, 25, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [Green Version]
- Ruas, J.L.; Berchner-Pfannschmidt, U.; Malik, S.; Gradin, K.; Fandrey, J.; Roeder, R.G.; Pereira, T.; Poellinger, L. Complex Regulation of the Transactivation Function of Hypoxia-inducible Factor-1α by Direct Interaction with Two Distinct Domains of the CREB-binding Protein/p300. J. Biol. Chem. 2010, 285, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Ruas, J.L.; Poellinger, L. Hypoxia-dependent activation of HIF into a transcriptional regulator. Semin. Cell Dev. Biol. 2005, 16, 514–522. [Google Scholar] [CrossRef]
- Ruas, J.; Poellinger, L.; Pereira, T. Role of CBP in regulating HIF-1-mediated activation of transcription. J. Cell Sci. 2005, 118, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- McNEILL, L.A.; Hewitson, K.S.; Claridge, T.D.; Seibel, J.F.; Horsfall, L.E.; Schofield, C.J. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the β-carbon of asparagine-803. Biochem. J. 2002, 367, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Carrero, P.; Okamoto, K.; Coumailleau, P.; O’Brien, S.; Tanaka, H.; Poellinger, L. Redox-Regulated Recruitment of the Transcriptional Coactivators CREB-Binding Protein and SRC-1 to Hypoxia-Inducible Factor 1α. Mol. Cell Biol. 2000, 20, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Chaika, N.V.; Gebregiworgis, T.; Lewallen, M.E.; Purohit, V.; Radhakrishnan, P.; Liu, X.; Zhang, B.; Mehla, K.; Brown, R.B.; Caffrey, T.; et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 13787–13792. [Google Scholar] [CrossRef] [Green Version]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A–dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Jeong, D.-W.; Park, J.-W.; Lee, K.-W.; Fukuda, J.; Chun, Y.-S. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun. Biol. 2020, 3, 638. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Michels, C.L.; Leung, M.-K.; Arany, Z.P.; Kung, A.L.; Livingston, D.M. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999, 13, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Freedman, S.J.; Sun, Z.-Y.J.; Kung, A.; France, D.S.; Wagner, G.; Eck, M.J. Structural basis for negative regulation of hypoxia-inducible factor-1α by CITED2. Nat. Struct. Mol. Biol. 2003, 10, 504–512. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Baek, J.H.; Semenza, G.L. Four-and-a-Half LIM Domain Proteins Inhibit Transactivation by Hypoxia-inducible Factor 1. J. Biol. Chem. 2012, 287, 6139–6149. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Shin, S.-H.; Shin, H.-W.; Chun, Y.-S.; Park, J.-W. Nuclear FGFR2 negatively regulates hypoxia-induced cell invasion in prostate cancer by interacting with HIF-1 and HIF-2. Sci. Rep. 2019, 9, 3480. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liu, X.; Mei, Z.; Wang, Z.; Xiao, W.; Ferreira, D.M.S.; Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Pereira, D.M.; et al. EAF2 Suppresses Hypoxia-Induced Factor 1α Transcriptional Activity by Disrupting Its Interaction with Coactivator CBP/p300. Mol. Cell Biol. 2014, 34, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Kim, Y.; Kim, I.S.; Kim, B.; Choi, H.J.; Lee, J.M.; Shin, H.R.; Kim, J.H.; Kim, J.-Y.; Seo, S.-B.; et al. Negative Regulation of Hypoxic Responses via Induced Reptin Methylation. Mol. Cell 2010, 39, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Kim, Y.; Bhin, J.; Shin, H.-J.R.; Nam, H.J.; Lee, S.H.; Yoon, J.-B.; Binda, O.; Gozani, O.; Hwang, D.; et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc. Natl. Acad. Sci. USA 2011, 108, 13510–13515. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367–E3376. [Google Scholar] [CrossRef] [Green Version]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.-I.; et al. Dynamic Change of Chromatin Conformation in Response to Hypoxia Enhances the Expression of GLUT3 (SLC2A3) by Cooperative Interaction of Hypoxia-Inducible Factor 1 and KDM3A. Mol. Cell Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Peng, K.; Li, M.; Qin, L.; Tong, Z.; Yan, J.; Shen, B.; Yu, C. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene 2017, 36, 3868–3877. [Google Scholar] [CrossRef]
- Mariani, C.J.; Vasanthakumar, A.; Madzo, J.; Yesilkanal, A.; Bhagat, T.; Yu, Y.; Verma, A. TET1-Mediated Hydroxymethylation Facilitates Hypoxic Gene Induction in Neuroblastoma. Cell Rep. 2014, 7, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-P.; Chen, H.-F.; Chen, S.-Y.; Cheng, W.-C.; Wang, H.-W.; Shen, Z.-J.; Song, C.; Teng, S.-C.; He, C.; Wu, K.-J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014, 15, 513. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Geng, H.; Xue, C.; Beer, T.M.; Qian, D.Z. Functional regulation of hypoxia inducible factor-1α by SET9 lysine methyltransferase. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, B.M.; Burrows, N.; Lobb, I.T.; Arnaiz, E.; Wit, N.; Bailey, P.S.J.; Jordon, L.H.; Lombardi, O.; Peñalver, A.; McCaffrey, J.; et al. The HIF complex recruits the histone methyltransferase SET1B to activate specific hypoxia-inducible genes. Nat. Genet. 2021, 53, 1022–1035. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, Y.; Tu, K.; Xu, Q.; Yang, Y.; Salman, S.; Le, N.; Lu, H.; Chen, C.; Zhu, Y.; et al. Histone citrullination by PADI4 is required for HIF-dependent transcriptional responses to hypoxia and tumor vascularization. Sci. Adv. 2021, 7, eabe3771. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, B.; Bao, L.; Jin, L.; Yang, M.; Peng, Y.; Kumar, A.; Wang, J.E.; Wang, C.; Zou, X.; et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Investig. 2018, 128, 1937–1955. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.; et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lu, H.; Chen, C.; Lyu, Y.; Cole, R.N.; Semenza, G.L. HIF-1 Interacts with TRIM28 and DNA-PK to release paused RNA polymerase II and activate target gene transcription in response to hypoxia. Nat. Commun. 2022, 13, 316. [Google Scholar] [CrossRef]
- Sena, J.A.; Wang, L.; Hu, C.-J.; Kim, S.-G.; Lee, B.; Kim, D.-H.; Kim, J.; Lee, S.; Lee, S.-K.; Lee, J.W. BRG1 and BRM Chromatin-Remodeling Complexes Regulate the Hypoxia Response by Acting as Coactivators for a Subset of Hypoxia-Inducible Transcription Factor Target Genes. Mol. Cell Biol. 2013, 33, 3849–3863. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Y.; Bao, L.; Zhang, B.; Wang, J.E.; Kumar, A.; Xing, C.; Wang, Y.; Luo, W. CHD4 Promotes Breast Cancer Progression as a Coactivator of Hypoxia-Inducible Factors. Cancer Res. 2020, 80, 3880–3891. [Google Scholar] [CrossRef]
- Koukoulas, K.; Giakountis, A.; Karagiota, A.; Samiotaki, M.; Panayotou, G.; Simos, G.; Mylonis, I. ERK signaling controls productive HIF-1 binding to chromatin and cancer cell adaptation to hypoxia through HIF-1α interaction with NPM1. Mol. Oncol. 2021, 15, 3468–3489. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-J.; Hsieh, Y.-J.; Cheng, W.-C.; Lin, C.-P.; Lin, Y.-S.; Yang, S.-F.; Chen, C.-C.; Izumiya, Y.; Yu, J.-S.; Kung, H.-J.; et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α–mediated glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Han, S.-N.; Zhang, J.-Y.; Dioletis, E.; Nemeth, B.T.; Pacher, P.; Feng, D.; Bataller, R.; Cabezas, J.; Stärkel, P.; et al. Digoxin Suppresses Pyruvate Kinase M2-Promoted HIF-1α Transactivation in Steatohepatitis. Cell Metab. 2018, 27, 339–350.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; Bosch, M.W.V.D.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Qiu, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Elser, M.; Borsig, L.; Hassa, P.O.; Erener, S.; Messner, S.; Valovka, T.; Keller, S.; Gassmann, M.; Hottiger, M.O. Poly(ADP-Ribose) Polymerase 1 Promotes Tumor Cell Survival by Coactivating Hypoxia-Inducible Factor-1–Dependent Gene Expression. Mol. Cancer Res. 2008, 6, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhou, A.-X.; Rouhi, P.; Uramoto, H.; Borén, J.; Cao, Y.; Pereira, T.; Akyürek, L.M.; Poellinger, L. Hypoxia-induced and calpain-dependent cleavage of filamin A regulates the hypoxic response. Proc. Natl. Acad. Sci. USA 2014, 111, 2560–2565. [Google Scholar] [CrossRef] [Green Version]
- Spiegelman, B.M.; Heinrich, R. Biological Control through Regulated Transcriptional Coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Yasinska, I.M.; Sumbayev, V.V. S-nitrosation of Cys-800 of HIF-1α protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Gradin, K.; Takasaki, C.; Fujii-Kuriyama, Y.; Sogawa, K. The Transcriptional Activation Function of the HIF-like Factor Requires Phosphorylation at a Conserved Threonine. J. Biol. Chem. 2002, 277, 23508–23514. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.-M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible Factor (HIF) Asparagine Hydroxylase Is Identical to Factor Inhibiting HIF (FIH) and Is Related to the Cupin Structural Family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020, 287, 3888–3906. [Google Scholar] [CrossRef]
- Torchia, J.; Glass, C.; Rosenfeld, M.G. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 1998, 10, 373–383. [Google Scholar] [CrossRef]
- Chen, H.; Lin, R.J.; Schiltz, R.; Chakravarti, D.; Nash, A.; Nagy, L.; Privalsky, M.L.; Nakatani, Y.; Evans, R.M. Nuclear Receptor Coactivator ACTR Is a Novel Histone Acetyltransferase and Forms a Multimeric Activation Complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.-C.; Heyman, A.R.; Rose, D.W.; Glass, C.K.; et al. A CBP Integrator Complex Mediates Transcriptional Activation and AP-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.P.; Ku, G.; Zhou, N.; Scully, R.; Livingston, D.M. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc. Natl. Acad. Sci. USA 1996, 93, 10626–10631. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Ballester, B.; Milara, J.; Cortijo, J. The role of mucin 1 in respiratory diseases. Eur. Respir. Rev. 2021, 30, 200149. [Google Scholar] [CrossRef]
- Aubert, S.; Fauquette, V.; Hémon, B.; Lepoivre, R.; Briez, N.; Bernard, D.; Van Seuningen, I.; Leroy, X.; Perrais, M. MUC1, a New Hypoxia Inducible Factor Target Gene, Is an Actor in Clear Renal Cell Carcinoma Tumor Progression. Cancer Res. 2009, 69, 5707–5715. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, D.; Zhou, Q.; Chen, T.; Ibe, J.C.F.; Raj, J.U.; Zhou, G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial–mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal. 2012, 24, 2396–2406. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Ai, J.; Habermacher, G.; Volpert, O.; Yang, X.; Zhang, A.-Y.; Hahn, J.; Cai, X.; Wang, Z. U19/Eaf2 Binds to and Stabilizes von Hippel-Lindau Protein. Cancer Res. 2009, 69, 2599–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, L.H.; Boussouar, F.; Boyd, K.; Xu, W.; Biesen, M.; Rehg, J.; Baudino, A.T.; Cleveland, J.L.; Brindle, P.K. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005, 24, 3846–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judes, G.; Rifaï, K.; Ngollo, M.; Daures, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Control of transcription by Pontin and Reptin. Trends Cell Biol. 2007, 17, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.-Q.; Houry, W.A. The Role of Pontin and Reptin in Cellular Physiology and Cancer Etiology. Front. Mol. Biosci. 2017, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic Stress Induces Dimethylated Histone H3 Lysine 9 through Histone Methyltransferase G9a in Mammalian Cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef] [Green Version]
- Gkotinakou, I.M.; Befani, C.; Samiotaki, M.; Panayotou, G.; Liakos, P. Novel HIF-2α interaction with Reptin52 impairs HIF-2 transcriptional activity and EPO secretion. Biochem. Biophys. Res. Commun. 2021, 557, 143–150. [Google Scholar] [CrossRef]
- Pollard, P.; Loenarz, C.; Mole, D.R.; McDonough, M.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The Histone Demethylases JMJD1A and JMJD2B Are Transcriptional Targets of Hypoxia-inducible Factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, X.W.Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Christophorou, M.A. The virtues and vices of protein citrullination. R. Soc. Open Sci. 2022, 9, 220125. [Google Scholar] [CrossRef]
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. Regulation of ZMYND8 to Treat Cancer. Molecules 2021, 26, 1083. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2017, 19, 262–274. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Bunch, H.; Zheng, X.; Burkholder, A.; Dillon, S.T.; Motola, S.; Birrane, G.; Ebmeier, C.C.; Levine, S.; Fargo, D.; Hu, G.; et al. TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat. Struct. Mol. Biol. 2014, 21, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Reid, X.J.; Low, J.K.; Mackay, J.P. A NuRD for all seasons. Trends Biochem. Sci. 2022, 48, 11–25. [Google Scholar] [CrossRef]
- López, D.J.; Rodríguez, J.A.; Bañuelos, S. Nucleophosmin, a multifunctional nucleolar organizer with a role in DNA repair. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140532. [Google Scholar] [CrossRef]
- Lin, J.; Kato, M.; Nagata, K.; Okuwaki, M. Efficient DNA binding of NF-κB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2016, 45, 3707–3723. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK Phosphorylation Sites and Their Role in the Localization and Activity of Hypoxia-inducible Factor-1α. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, X.; Sejas, D.P.; Bagby, G.C.; Pang, Q. Hypoxia-induced Nucleophosmin Protects Cell Death through Inhibition of p53. J. Biol. Chem. 2004, 279, 41275–41279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiota, A.; Kourti, M.; Simos, G.; Mylonis, I. HIF-1α-derived cell-penetrating peptides inhibit ERK-dependent activation of HIF-1 and trigger apoptosis of cancer cells under hypoxia. Cell Mol. Life Sci. 2018, 76, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.-K.; Spille, J.-H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM 2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.O.; Del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.C.; Shibuya, M. A variant of nuclear localization signal of bipartite-type is required for the nuclear translocation of hypoxia inducible factors (1α, 2α and 3α). Oncogene 2001, 20, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent Nuclear Export Signal Mediates Regulation of Hypoxia-inducible Factor-1α by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Daly, L.; Mennerich, D.; Kietzmann, T.; Sée, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int. J. Mol. Sci. 2020, 22, 268. [Google Scholar] [CrossRef]
- Zong, W.; Gong, Y.; Sun, W.; Li, T.; Wang, Z.-Q. PARP1: Liaison of Chromatin Remodeling and Transcription. Cancers 2022, 14, 4162. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, X.; An, H.; Lv, Y.; Liu, X. The function and pathogenic mechanism of filamin A. Gene 2021, 784, 145575. [Google Scholar] [CrossRef]
- Tanimoto, K.; Tsuchihara, K.; Kanai, A.; Arauchi, T.; Esumi, H.; Suzuki, Y.; Sugano, S. Genome-wide identification and annotation of HIF-1α binding sites in two cell lines using massively parallel sequencing. HUGO J. 2010, 4, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Hulsen, T. DeepVenn—A web application for the creation of area-proportional Venn diagrams using the deep learning framework. arXiv, 2022; arXiv:2210.04597v1. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Contributors. SciPy 1.0 Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]

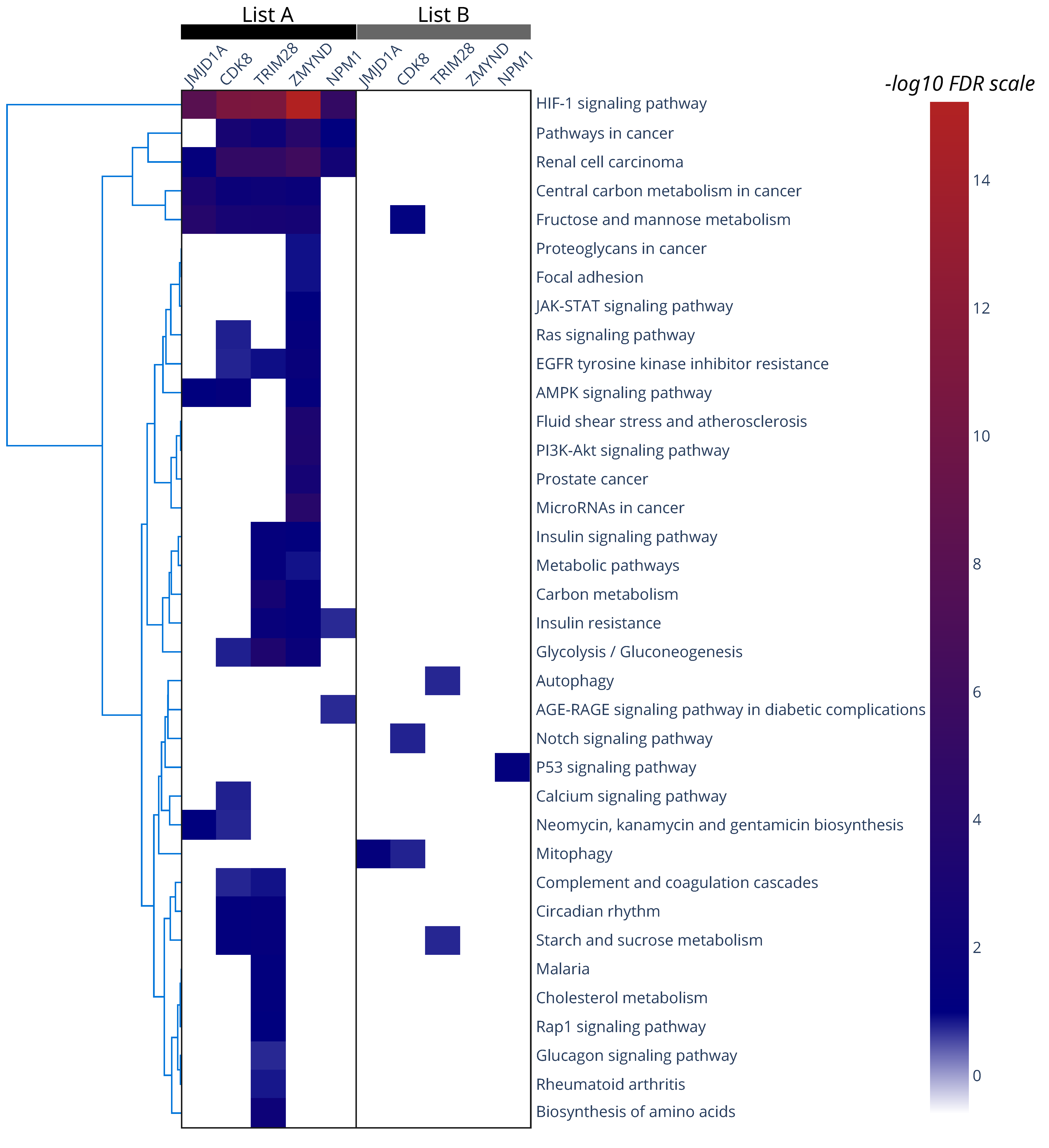

| HIF-1 Co-Activator | Ref. | Number of | ||
|---|---|---|---|---|
| Co-Activator- Dependent Genes | Common Genes with List A (83 Genes) | Common Genes with List B (109 Genes) | ||
| ZMYND8 # | [53] | 603 | 45 * | 62 * |
| CDK8 | [54] | 168 | 34 * | 31 * |
| TRIM 28 | [55] | 1101 | 34 * | 42 * |
| NPM1 # | [58] | 436 | 19 * | 12 * |
| JMJD1A # | [46] | 224 | 15 * | 13 * |
| TIP60 | [44] | 131 | 9 * | 5 |
| TET1 # | [49] | 1044 | 16 | 10 |
| Reptin | [42] | 35 | 5 | 4 |
| Pontin | [43] | 66 | 2 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yfantis, A.; Mylonis, I.; Chachami, G.; Nikolaidis, M.; Amoutzias, G.D.; Paraskeva, E.; Simos, G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells 2023, 12, 798. https://doi.org/10.3390/cells12050798
Yfantis A, Mylonis I, Chachami G, Nikolaidis M, Amoutzias GD, Paraskeva E, Simos G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells. 2023; 12(5):798. https://doi.org/10.3390/cells12050798
Chicago/Turabian StyleYfantis, Angelos, Ilias Mylonis, Georgia Chachami, Marios Nikolaidis, Grigorios D. Amoutzias, Efrosyni Paraskeva, and George Simos. 2023. "Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators" Cells 12, no. 5: 798. https://doi.org/10.3390/cells12050798
APA StyleYfantis, A., Mylonis, I., Chachami, G., Nikolaidis, M., Amoutzias, G. D., Paraskeva, E., & Simos, G. (2023). Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells, 12(5), 798. https://doi.org/10.3390/cells12050798