
Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance



{kind=link}
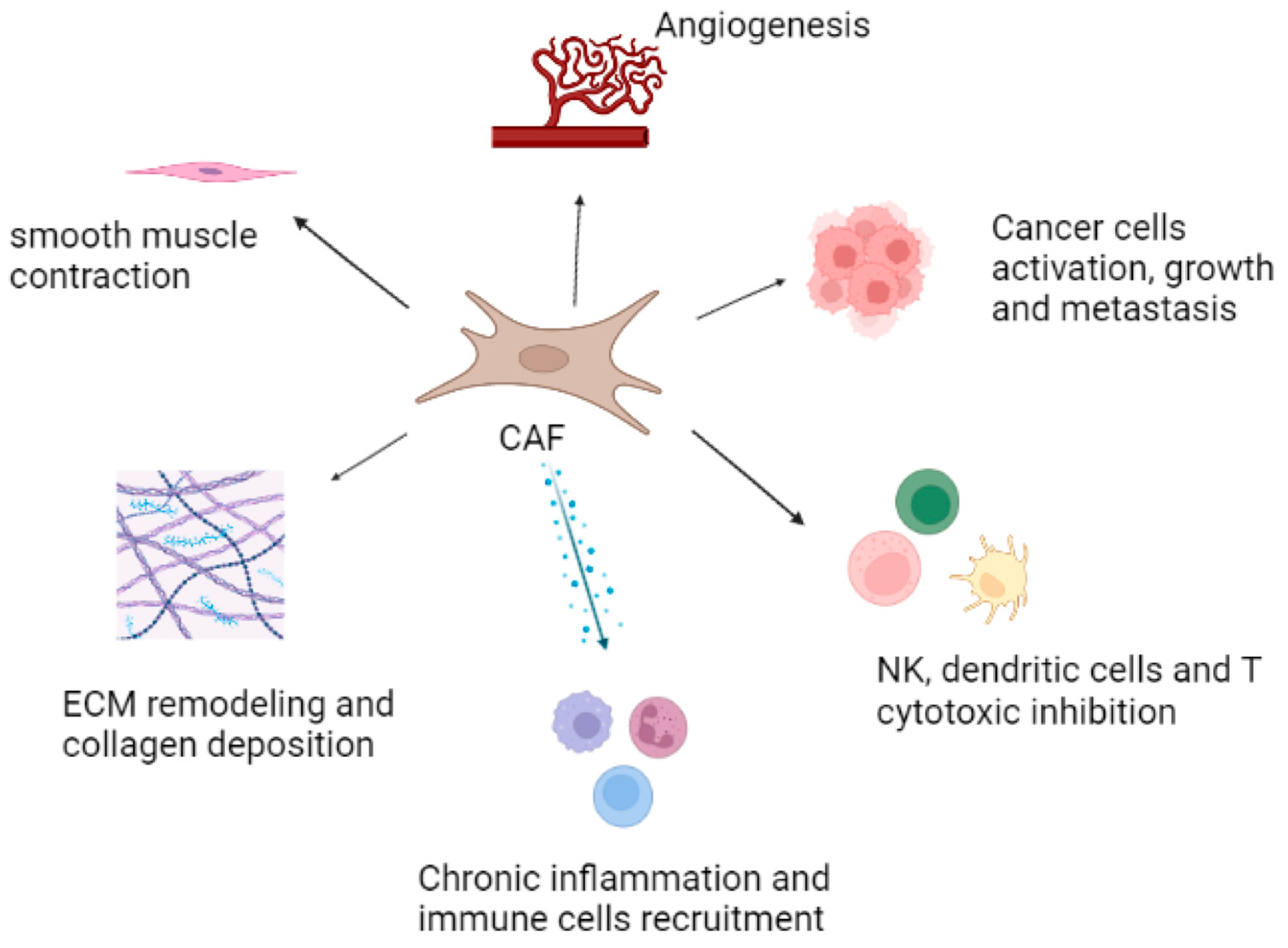{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Prostate Fibroblasts
3. Prostate Cancer-Associated Fibroblasts (CAFs)
4. Potential Sources of CAFs and Their Heterogeneity in Prostate Cancer
5. Role of CAFs in the Prostate Tumor Microenvironment and Cancer Progression
6. CAFs and Prostate Tumor Progression
7. Prostate CAFs and Therapeutic Resistance
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen-deprivation therapy |
| apCAFs | Antigen-presenting cancer-associated fibroblasts |
| AR | Androgen receptor |
| BC | Breast carcinoma |
| CAFs | Cancer-associated fibroblasts |
| DKK3 | Dickkopf-3 |
| ECM | Extracellular matrix |
| FAPIs | FAP inhibitor |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FlnA | Filamin A |
| ICIs | Immune checkpoint inhibitors |
| IGFBP | Insulin-like growth factor binding protein |
| LMO2 | LIM domain only 2 |
| mCRPC | Metastatic castration-resistant prostate cancer |
| MDSCs | Myeloid-derived suppressor cells |
| MMPs | Matrix-metalloproteinases |
| MSCs | Mesenchymal stem cells |
| NKs | Natural killers |
| PCa | Prostate cancer |
| PD-1 | Programmed cell death protein 1 |
| PDAC | Pancreatic adenocarcinoma |
| PDL-1 | Programmed cell death ligand 1 |
| ROS | Reactive oxygen species |
| TGF-β | Tumor growth factor β |
| TIMPs | Tissue inhibitors of matrix metalloproteinases |
| TME | Tumor microenvironment |
| VEGFA | Vascular endothelial growth factor A |
| YAP1 | Yes-associated protein 1 |
References
- ChallaSivaKanaka, S.; Vickman, R.E.; Kakarla, M.; Hayward, S.W.; Franco, O.E. Fibroblast heterogeneity in prostate carcinogenesis. Cancer Lett. 2022, 525, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and prevention of prostate cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Grozescu, T.; Popa, F. Prostate cancer between prognosis and adequate/proper therapy. J. Med. Life 2017, 10, 5. [Google Scholar]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of advanced prostate cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Howlader, N.N.A.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review 1975–2014; National Cancer Institute: Bethesda, MD, USA, 2018. Available online: https://seer.cancer.gov/csr/1975_2014/ (accessed on 2 March 2023).
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Lara, P.N.; Burch, P.A.; Kohli, M.; Raghavan, D. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. New Engl. J. Med. 2004, 8, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Qu, F.; Xie, W.; Nakabayashi, M.; Zhang, H.; Jeong, S.H.; Wang, X.; Komura, K.; Sweeney, C.J.; Sartor, O.; Lee, G.-S.M.; et al. Association of AR-V7 and prostate-specific antigen RNA levels in blood with efficacy of abiraterone acetate and enzalutamide treatment in men with prostate cancer. Clin. Cancer Res. 2017, 23, 726–734. [Google Scholar] [CrossRef]
- Lynch, H.T.; Kosoko-Lasaki, O.; Leslie, S.W.; Rendell, M.; Shaw, T.; Snyder, C.; D’Amico, A.V.; Buxbaum, S.; Isaacs, W.B.; Loeb, S.; et al. Screening for familial and hereditary prostate cancer. Int. J. Cancer 2016, 138, 2579–2591. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. New Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Hjelmborg, J.B.; Scheike, T.; Holst, K.; Skytthe, A.; Penney, K.L.; Graff, R.E.; Pukkala, E.; Christensen, K.; Adami, H.-O.; Holm, N.V.; et al. The heritability of prostate cancer in the Nordic twin study of cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Kote-Jarai, Z.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Castro, E.; Mahmud, N.; Guy, M.; Edwards, S.; O’Brien, L.; Sawyer, E.; et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br. J. Cancer 2011, 105, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamornlert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Tryggvadóttir, L.; Vidarsdóttir, L.; Thorgeirsson, T.; Jonasson, J.G.; Ólafsdóttir, E.J.; Ólafsdóttir, G.H.; Rafnar, T.; Thorlacius, S.; Jonsson, E.; Eyfjord, J.E.; et al. Prostate cancer progression and survival in BRCA2 mutation carriers. JNCI J. Natl. Cancer Inst. 2007, 99, 929–935. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yoshimoto, M.; Cutz, J.-C.; Nuin, P.A.; Joshua, A.M.; Bayani, J.; Evans, A.J.; Zielenska, M.; Squire, J.A. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet. Cytogenet. 2006, 169, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Koo, K.M.; Mainwaring, P.N.; Tomlins, S.A.; Trau, M. Merging new-age biomarkers and nanodiagnostics for precision prostate cancer management. Nat. Rev. Urol. 2019, 16, 302–317. [Google Scholar] [CrossRef]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Mosquera, J.M.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef]
- Reig, Ò.; Marín-Aguilera, M.; Carrera, G.; Jiménez, N.; Paré, L.; García-Recio, S.; Gaba, L.; Pereira, M.V.; Fernández, P.; Prat, A.; et al. TMPRSS2-ERG in blood and docetaxel resistance in metastatic castration-resistant prostate cancer. Eur. Urol. 2016, 70, 709–713. [Google Scholar] [CrossRef]
- Zhao, S.G.; Lehrer, J.; Chang, S.L.; Das, R.; Erho, N.; Liu, Y.; Sjöström, M.; Den, R.B.; Freedland, S.J.; Klein, E.A.; et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. JNCI J. Natl. Cancer Inst. 2018, 111, 301–310. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zeng, H.; Chen, Y. Emerging nano drug delivery systems targeting cancer-associated fibroblasts for improved antitumor effect and tumor drug penetration. Mol. Pharm. 2020, 17, 1028–1048. [Google Scholar] [CrossRef] [PubMed]
- El-Alfy, M.; Pelletier, G.; Hermo, L.S.; Labrie, F. Unique features of the basal cells of human prostate epithelium. Microsc. Res. Tech. 2000, 51, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Hayward, S.W. Targeting the tumor stroma as a novel therapeutic approach for prostate cancer. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 65, pp. 267–313. ISBN 978-0-12-397927-8. [Google Scholar] [CrossRef]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases: The myofibroblast. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Buechler, M.B.; Turley, S.J. A short field guide to fibroblast function in immunity. Semin. Immunol. 2018, 35, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.-C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef]
- Yip, W.L. Influence of oxygen on wound healing. Int. Wound J. 2014, 12, 620–624. [Google Scholar] [CrossRef]
- Kan, C.; Abe, M.; Yamanaka, M.; Ishikawa, O. Hypoxia-induced increase of matrix metalloproteinase-1 synthesis is not restored by reoxygenation in a three-dimensional culture of human dermal fibroblasts. J. Dermatol. Sci. 2003, 32, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Krušlin, B.; Ulamec, M.; Tomas, D. Prostate cancer stroma: An important factor in cancer growth and progression. Bosn. J. Basic Med. Sci. 2015, 15, 1. [Google Scholar] [CrossRef]
- Begley, L.A.; Kasina, S.; MacDonald, J.; Macoska, J.A. The inflammatory microenvironment of the aging prostate facilitates cellular proliferation and hypertrophy. Cytokine 2008, 43, 194–199. [Google Scholar] [CrossRef]
- Levesque, C.; Nelson, P.S. Cellular constituents of the prostate stroma: Key contributors to prostate cancer progression and therapy resistance. Cold Spring Harb. Perspect. Med. 2017, 8, a030510. [Google Scholar] [CrossRef]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-associated fibroblasts modify the response of prostate cancer cells to androgen and anti-androgens in three-dimensional spheroid culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [PubMed]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular matrices and cancer-associated fibroblasts: Targets for cancer diagnosis and therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Chiarugi, P.; Paoli, P.; Cirri, P. Tumor microenvironment and metabolism in prostate cancer. Semin. Oncol. 2014, 41, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Sun, D.-Y.; Wu, J.-Q.; He, Z.-H.; He, M.-F.; Sun, H.-B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Guyot, C.; Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef]
- Cat, B.; Stuhlmann, D.; Steinbrenner, H.; Alili, L.; Holtkötter, O.; Sies, H.; Brenneisen, P. Enhancement of tumor invasion depends on transdifferentiation of skin fibroblasts mediated by reactive oxygen species. J. Cell Sci. 2006, 119, 2727–2738. [Google Scholar] [CrossRef]
- Shen, T.; Li, Y.; Zhu, S.; Yu, J.; Zhang, B.; Chen, X.; Zhang, Z.; Ma, Y.; Niu, Y.; Shang, Z. YAP1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 36. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.; Dembinski, J.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef]
- Krtolica, A.; Campisi, J. Cancer and aging: A model for the cancer promoting effects of the aging stroma. Int. J. Biochem. Cell Biol. 2002, 34, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef]
- Laurent, V.; Toulet, A.; Attané, C.; Milhas, D.; Dauvillier, S.; Zaidi, F.; Clement, E.; Cinato, M.; Le Gonidec, S.; Guérard, A.; et al. Periprostatic adipose tissue favors prostate cancer cell invasion in an obesity-dependent manner: Role of oxidative stress. Mol. Cancer Res. 2019, 17, 821–835. [Google Scholar] [CrossRef]
- Ribeiro, R.; Monteiro, C.; Cunha, V.; Oliveira, M.J.; Freitas, M.; Fraga, A.; Príncipe, P.; Lobato, C.; Lobo, F.; Morais, A.; et al. Human periprostatic adipose tissue promotes prostate cancer aggressiveness in vitro. J. Exp. Clin. Cancer Res. 2012, 31, 32. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Deasy, S.K.; Erez, N. A glitch in the matrix: Organ-specific matrisomes in metastatic niches. Trends Cell Biol. 2022, 32, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X.; et al. Proteomic profiling of human prostate cancer-associated fibroblasts (CAF) reveals LOXL2-dependent regulation of the tumor microenvironment. Mol. Cell Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef]
- Kerdidani, D.; Aerakis, E.; Verrou, K.-M.; Angelidis, I.; Douka, K.; Maniou, M.-A.; Stamoulis, P.; Goudevenou, K.; Prados, A.; Tzaferis, C.; et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J. Exp. Med. 2022, 219, e20210815. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-De-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2013, 33, 2423–2431. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Ma, J.P.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2020, 80, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Sala, M.; Ros, M.; Saltel, F. A complex and evolutive character: Two face aspects of ECM in tumor progression. Front. Oncol. 2020, 10, 1620. [Google Scholar] [CrossRef] [PubMed]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nature 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, B.; Zhang, L.; Dang, T.; Rowley, D.; Ittmann, M.; Xin, L. Jagged1 upregulation in prostate epithelial cells promotes formation of reactive stroma in the Pten null mouse model for prostate cancer. Oncogene 2017, 36, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S., II; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: In-duction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar] [PubMed]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical signaling of the extracellular matrix in epithelial-mesenchymal transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, A.; Jacobi, A.; Lawrence, M.G.; Risbridger, G.P.; Frydenberg, M.; Williams, E.D.; Vela, I.; Hutmacher, D.W.; Bray, L.J.; Taubenberger, A. Cancer-associated fibroblasts of the prostate promote a compliant and more invasive phenotype in benign prostate epithelial cells. Mater. Today Bio 2020, 8, 100073. [Google Scholar] [CrossRef]
- Escaff, S.; Fernández, J.M.; González, L.O.; Suárez, A.; González-Reyes, S.; González, J.M.; Vizoso, F.J. Study of matrix metalloproteinases and their inhibitors in prostate cancer. Br. J. Cancer 2010, 102, 922–929. [Google Scholar] [CrossRef]
- Fernandez-Gomez, J.; Escaf, S.; Gonzalez, L.-O.; Suarez, A.; Gonzalez-Reyes, S.; González, J.; Miranda, O.; Vizoso, F. Relationship between metalloprotease expression in tumour and stromal cells and aggressive behaviour in prostate carcinoma: Simultaneous high-throughput study of multiple metalloproteases and their inhibitors using tissue array analysis of radical prostatectomy samples. Scand. J. Urol. Nephrol. 2011, 45, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef]
- Wood, M.; Fudge, K.; Mohler, J.L.; Frost, A.R.; Garcia, F.; Wang, M.; Stearns, M.E. In situ hybridization studies of metalloproteinases 2 and 9 and TIMP-1 and TIMP-2 expression in human prostate cancer. Clin. Exp. Metastasis 1997, 15, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Al-Shareef, Z.; Gorroño-Etxebarria, I.; Atkins, S.; Turrell, F.; Chhetri, J.; Bengoa-Vergniory, N.; Zenzmaier, C.; Berger, P.; Waxman, J.; et al. Dickkopf-3 regulates prostate epithelial cell acinar morphogenesis and prostate cancer cell invasion by limiting TGF-β-dependent activation of matrix metalloproteases. Carcinogenesis 2016, 37, 18–29. [Google Scholar] [CrossRef]
- Al Shareef, Z.; Kardooni, H.; Murillo-Garzón, V.; Domenici, G.; Stylianakis, E.; Steel, J.H.; Rabano, M.; Gorroño-Etxebarria, I.; Zabalza, I.; Vivanco, M.D.; et al. Protective effect of stromal Dickkopf-3 in prostate cancer: Opposing roles for TGFBI and ECM-1. Oncogene 2018, 37, 5305–5324. [Google Scholar] [CrossRef]
- Castoria, G.; Giovannelli, P.; Di Donato, M.; Ciociola, A.; Hayashi, R.; Bernal, F.; Appella, E.; Auricchio, F.; Migliaccio, A. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014, 5, e1548. [Google Scholar] [CrossRef]
- Castoria, G.; Lombardi, M.; Barone, M.V.; Bilancio, A.; Di Domenico, M.; Bottero, D.; Vitale, F.; Migliaccio, A.; Auricchio, F. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J. Cell Biol. 2003, 161, 547–556. [Google Scholar] [CrossRef]
- Di Donato, M.; Zamagni, A.; Galasso, G.; Di Zazzo, E.; Giovannelli, P.; Barone, M.V.; Zanoni, M.; Gunelli, R.; Costantini, M.; Auricchio, F.; et al. The androgen receptor/filamin A complex as a target in prostate cancer microenvironment. Cell Death Dis. 2021, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kajiwara, S.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Mizokami, A.; Sugimura, Y. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate 2018, 78, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, L.; Lin, H.-K.; Kan, P.-Y.; Xie, S.; Tsai, M.-Y.; Wang, P.-H.; Chen, Y.-T.; Chang, C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem. Biophys. Res. Commun. 2003, 305, 462–469. [Google Scholar] [CrossRef]
- Leach, D.A.; Buchanan, G. Stromal androgen receptor in prostate cancer development and progression. Cancers 2017, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Cioni, B.; Nevedomskaya, E.; Melis, M.H.M.; Van Burgsteden, J.; Stelloo, S.; Hodel, E.; Spinozzi, D.; De Jong, J.; Van Der Poel, H.; De Boer, J.P.; et al. Loss of androgen receptor signaling in prostate cancer-associated fibroblasts (CAFs) promotes CCL2- and CXCL8-mediated cancer cell migration. Mol. Oncol. 2018, 12, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.; Östman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front. Biosci. 2018, 23, 626–639. [Google Scholar] [CrossRef]
- Li, D.; Xia, L.; Huang, P.; Wang, Z.; Guo, Q.; Huang, C.; Leng, W.; Qin, S. Cancer-associated fibroblast-secreted IGFBP7 promotes gastric cancer by enhancing tumor associated macrophage infiltration via FGF2/FGFR1/PI3K/AKT axis. Cell Death Discov. 2023, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. -Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Dakhova, O.; Creighton, C.J.; Ittmann, M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013, 73, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Labanca, E.; Yang, J.; Shepherd, P.D.; Wan, X.; Starbuck, M.W.; Guerra, L.D.; Anselmino, N.; Bizzotto, J.A.; Dong, J.; Chinnaiyan, A.M.; et al. Fibroblast growth factor receptor 1 drives the metastatic progression of prostate cancer. Eur. Urol. Oncol. 2021, 5, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Y.-Y.; Li, D.; Wang, C.; Wang, S.-Y.; Shao, S.-H.; Zhu, Z.-Y.; Zhao, J.; Zhang, Y.; Ruan, Y.; et al. LMO2 upregulation due to AR deactivation in cancer-associated fibroblasts induces non-cell-autonomous growth of prostate cancer after androgen deprivation. Cancer Lett. 2021, 503, 138–150. [Google Scholar] [CrossRef]
- Liow, E.; Howard, N.; Jung, C.-H.; Pope, B.; Campbell, B.K.; Nguyen, A.; Kerger, M.; Ruddle, J.B.; Anton, A.; Thomas, B.; et al. Phase 2 study of neoadjuvant FGFR inhibition and androgen deprivation therapy prior to prostatectomy. Clin. Genitourin. Cancer 2022, 20, 452–458. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of theranostics: An outlook on precision oncology in nuclear medicine. J. Nucl. Med. 2019, 60, 13S–19S. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedeschi, M.; Marino, N.; Cavassi, E.; Piccinini, F.; Tesei, A. Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells 2023, 12, 802. https://doi.org/10.3390/cells12050802
Bedeschi M, Marino N, Cavassi E, Piccinini F, Tesei A. Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells. 2023; 12(5):802. https://doi.org/10.3390/cells12050802
Chicago/Turabian StyleBedeschi, Martina, Noemi Marino, Elena Cavassi, Filippo Piccinini, and Anna Tesei. 2023. "Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance" Cells 12, no. 5: 802. https://doi.org/10.3390/cells12050802
APA StyleBedeschi, M., Marino, N., Cavassi, E., Piccinini, F., & Tesei, A. (2023). Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells, 12(5), 802. https://doi.org/10.3390/cells12050802