
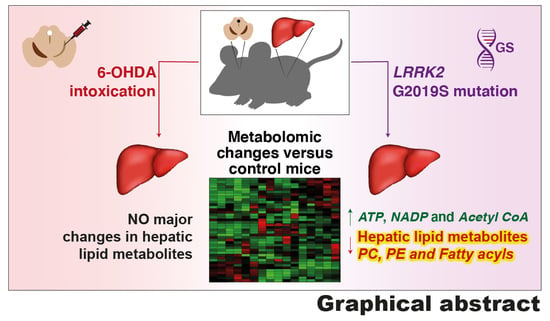
Changes in Liver Lipidomic Profile in G2019S-LRRK2 Mouse Model of Parkinson’s Disease

 ,
,  , , , ,
, , , ,  ,
,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Metabolic Changes Observed in Nerve Cells of Genetic and Acute Intoxication Models of PD
3.2. Metabolic Changes Observed in the Hepatocytes of Genetic and Acute Intoxication Models of PD
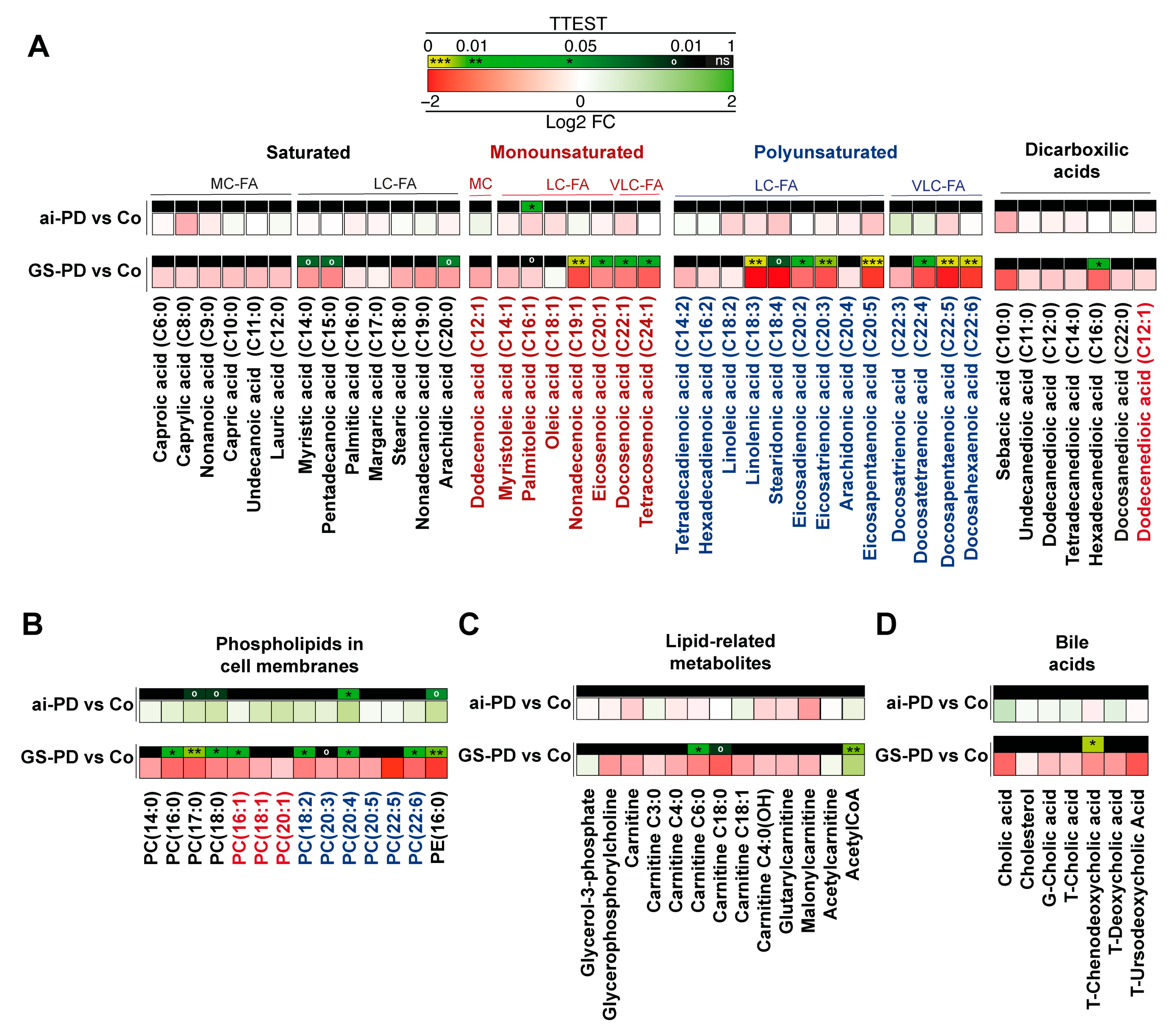
3.3. Changes in Lipid Metabolites in Genetic or Acute Intoxication PD Models Liver
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Karikari, T.K.; Charway-Felli, A.; Hoglund, K.; Blennow, K.; Zetterberg, H. Commentary: Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Front. Neurol. 2018, 9, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit Mechanisms of Parkinson’s Disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell. Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [Green Version]
- Vidovic, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef]
- Reyes, J.F.; Ekmark-Lewen, S.; Perdiki, M.; Klingstedt, T.; Hoffmann, A.; Wiechec, E.; Nilsson, P.; Nilsson, K.P.R.; Alafuzoff, I.; Ingelsson, M.; et al. Accumulation of alpha-synuclein within the liver, potential role in the clearance of brain pathology associated with Parkinson’s disease. Acta Neuropathol. Commun. 2021, 9, 46. [Google Scholar] [CrossRef]
- Sato, S.; Mizuno, Y.; Hattori, N. Urinary 8-hydroxydeoxyguanosine levels as a biomarker for progression of Parkinson disease. Neurology 2005, 64, 1081–1083. [Google Scholar] [CrossRef]
- Ishizakai, M.; Yoshida, K.; Nishimoto, N.; Saleh, A.M.; Ishii, C.; Handa, H.; Ogawara, H.; Nagamine, T.; Murakami, M.; Murakami, H. Urinary 8-hydroxy-2’-deoxyguanosin (8-OHdG) in patients with chronic liver diseases. Rinsho Byori 2004, 52, 732–736. [Google Scholar] [PubMed]
- Apetauerova, D.; Hildebrand, P.; Scala, S.; Zani, J.W.; Lipert, L.; Clark, E.; Fennell, T.; Gordon, F.D. A Prospective Study of the Prevalence of Parkinsonism in Patients With Liver Cirrhosis. Hepatol. Commun. 2021, 5, 323–333. [Google Scholar] [CrossRef]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.; Vinikoor-Imler, L.; Nassan, F.L.; Shirvan, J.; Lally, C.; Dam, T.; Maserejian, N. Prevalence of ten LRRK2 variants in Parkinson’s disease: A comprehensive review. Park. Relat. Disord. 2022, 98, 103–113. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.; Bravo-San Pedro, J.M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Rodriguez-Arribas, M.; Climent, V.; Aiastui, A.; Lopez de Munain, A.; Fuentes, J.M.; Gonzalez-Polo, R.A. G2019S LRRK2 mutant fibroblasts from Parkinson’s disease patients show increased sensitivity to neurotoxin 1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology 2014, 324, 1–9. [Google Scholar] [CrossRef]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; Lopez de Maturana, R.; Sanchez-Pernaute, R.; Lopez de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Cookson, M.R. LRRK2 recruitment, activity, and function in organelles. FEBS J. 2022, 289, 6871–6890. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Le, W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maetzler, W.; Schmid, S.P.; Wurster, I.; Liepelt, I.; Gaenslen, A.; Gasser, T.; Berg, D. Reduced but not oxidized cerebrospinal fluid glutathione levels are lowered in Lewy body diseases. Mov. Disord. 2011, 26, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Song, W.; Chen, K.; Chen, X.; Zheng, Z.; Cao, B.; Huang, R.; Zhao, B.; Wu, Y.; Shang, H.F. The serum lipid profile of Parkinson’s disease patients: A study from China. Int. J. Neurosci. 2015, 125, 838–844. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.F.; Meng, N.; Tang, Z.; Chua, K.K.; Chen, L.L.; Song, J.X.; Mok, V.C.; Xie, L.X.; Li, M.; et al. LC-MS-based urinary metabolite signatures in idiopathic Parkinson’s disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Zheng, W.; He, Y.; Tang, W.; Wei, X.; He, R.; Huang, W.; Su, Y.; Huang, Y.; Zhou, H.; et al. Gut microbiota in patients with Parkinson’s disease in southern China. Park. Relat. Disord. 2018, 53, 82–88. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X.; Zhao, L.; Yang, C.; Pan, L.; Li, C.; Liu, K.; Bai, G.; Gao, H.; Yan, Z. Metabolic Disturbances in the Striatum and Substantia Nigra in the Onset and Progression of MPTP-Induced Parkinsonism Model. Front. Neurosci. 2018, 12, 90. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Aguilar-Morante, D.; Hernandez-Encinas, E.; Alonso-Gil, S.; Gil, C.; Martinez, A.; Santos, A.; Perez-Castillo, A. Silencing phosphodiesterase 7B gene by lentiviral-shRNA interference attenuates neurodegeneration and motor deficits in hemiparkinsonian mice. Neurobiol. Aging 2015, 36, 1160–1173. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Ramonet, D.; Daher, J.P.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 2011, 6, e18568. [Google Scholar] [CrossRef] [Green Version]
- Tozzi, A.; Tantucci, M.; Marchi, S.; Mazzocchetti, P.; Morari, M.; Pinton, P.; Mancini, A.; Calabresi, P. Dopamine D2 receptor-mediated neuroprotection in a G2019S Lrrk2 genetic model of Parkinson’s disease. Cell Death Dis. 2018, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Vairetti, M.; Ferrigno, A.; Rizzo, V.; Ambrosi, G.; Bianchi, A.; Richelmi, P.; Blandini, F.; Armentero, M.T. Impaired hepatic function and central dopaminergic denervation in a rodent model of Parkinson’s disease: A self-perpetuating crosstalk? Biochim. Biophys. Acta 2012, 1822, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ni, H.M.; Ding, Y.; Ding, W.X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.; Berg, D.; Bosy-Westphal, A.; Schaeffer, E. Parkinson’s Disease and Sugar Intake-Reasons for and Consequences of a Still Unclear Craving. Nutrients 2022, 14, 3240. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Tiberi, M.; Matteocci, A.; Fazio, F.; Siffeti, H.; Saracini, S.; Mercuri, N.B.; Sancesario, G. Lipidomics of Bioactive Lipids in Alzheimer’s and Parkinson’s Diseases: Where Are We? Int. J. Mol. Sci. 2022, 23, 6235. [Google Scholar] [CrossRef] [PubMed]
- Vendruscolo, M. Lipid Homeostasis and Its Links With Protein Misfolding Diseases. Front. Mol. Neurosci. 2022, 15, 829291. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [Green Version]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Gomez-Sanchez, R.; Niso-Santano, M.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; Lopez de Maturana, R.; Sanchez-Pernaute, R.; Lopez de Munain, A.; et al. The MAPK1/3 pathway is essential for the deregulation of autophagy observed in G2019S LRRK2 mutant fibroblasts. Autophagy 2012, 8, 1537–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, N.; Ito, Y.; Otsuki, Y. Triggering of Parkin Mitochondrial Translocation in Mitophagy: Implications for Liver Diseases. Front. Pharmacol. 2016, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Gomes, F.A.; Flores, R.A.; Bruxel, M.A.; da Silva, F.N.; Moreira, E.L.G.; Zoccal, D.B.; Prediger, R.D.; Rafacho, A. Glucose Homeostasis Is Not Affected in a Murine Model of Parkinson’s Disease Induced by 6-OHDA. Front. Neurosci. 2018, 12, 1020. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.W.; Peng, Y.J.; Lin, Y.Y.; Mersmann, H.J.; Ding, S.T. LRRK2 Regulates CPT1A to Promote beta-Oxidation in HepG2 Cells. Molecules 2020, 25, 4122. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.A.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE 2013, 8, e80705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galper, J.; Kim, W.S.; Dzamko, N. LRRK2 and Lipid Pathways: Implications for Parkinson’s Disease. Biomolecules 2022, 12, 1597. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.S.; Rodriguez-Arribas, M.; Martinez-Chacon, G.; Uribe-Carretero, E.; Gomez-Sanchez, R.; Aiastui, A.; Lopez de Munain, A.; Bravo-San Pedro, J.M.; Niso-Santano, M.; Gonzalez-Polo, R.A.; et al. Acetylome in Human Fibroblasts From Parkinson’s Disease Patients. Front. Cell Neurosci. 2018, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodriguez-Arribas, M.; Gomez-Sanchez, R.; Martinez-Chacon, G.; Uribe-Carretero, E.; Navarro-Garcia, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wang, J.; Li, M.; Liu, Q.; Wei, D.; Yang, M.; Kong, L. (1)H NMR-based metabolomics study on a goldfish model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Chem. Biol. Interact. 2014, 223, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Novello, S.; Arcuri, L.; Dovero, S.; Dutheil, N.; Shimshek, D.R.; Bezard, E.; Morari, M. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol. Dis. 2018, 120, 21–33. [Google Scholar] [CrossRef]
- O’Leary, E.I.; Jiang, Z.; Strub, M.P.; Lee, J.C. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated alpha-synuclein. J. Biol. Chem. 2018, 293, 11195–11205. [Google Scholar] [CrossRef] [Green Version]
- Broersen, K.; Ruiperez, V.; Davletov, B. Structural and Aggregation Properties of Alpha-Synuclein Linked to Phospholipase A2 Action. Protein Pept. Lett. 2018, 25, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Custodia, A.; Aramburu-Nunez, M.; Correa-Paz, C.; Posado-Fernandez, A.; Gomez-Larrauri, A.; Castillo, J.; Gomez-Munoz, A.; Sobrino, T.; Ouro, A. Ceramide Metabolism and Parkinson’s Disease-Therapeutic Targets. Biomolecules 2021, 11, 945. [Google Scholar] [CrossRef]
- Esfandiary, A.; Finkelstein, D.I.; Voelcker, N.H.; Rudd, D. Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery. Cells 2022, 11, 1353. [Google Scholar] [CrossRef] [PubMed]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corral Nieto, Y.; Yakhine-Diop, S.M.S.; Moreno-Cruz, P.; Manrique García, L.; Gabrielly Pereira, A.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Durand, S.; et al. Changes in Liver Lipidomic Profile in G2019S-LRRK2 Mouse Model of Parkinson’s Disease. Cells 2023, 12, 806. https://doi.org/10.3390/cells12050806
Corral Nieto Y, Yakhine-Diop SMS, Moreno-Cruz P, Manrique García L, Gabrielly Pereira A, Morales-García JA, Niso-Santano M, González-Polo RA, Uribe-Carretero E, Durand S, et al. Changes in Liver Lipidomic Profile in G2019S-LRRK2 Mouse Model of Parkinson’s Disease. Cells. 2023; 12(5):806. https://doi.org/10.3390/cells12050806
Chicago/Turabian StyleCorral Nieto, Yaiza, Sokhna M. S. Yakhine-Diop, Paula Moreno-Cruz, Laura Manrique García, Amanda Gabrielly Pereira, José A. Morales-García, Mireia Niso-Santano, Rosa A. González-Polo, Elisabet Uribe-Carretero, Sylvère Durand, and et al. 2023. "Changes in Liver Lipidomic Profile in G2019S-LRRK2 Mouse Model of Parkinson’s Disease" Cells 12, no. 5: 806. https://doi.org/10.3390/cells12050806
APA StyleCorral Nieto, Y., Yakhine-Diop, S. M. S., Moreno-Cruz, P., Manrique García, L., Gabrielly Pereira, A., Morales-García, J. A., Niso-Santano, M., González-Polo, R. A., Uribe-Carretero, E., Durand, S., Maiuri, M. C., Paredes-Barquero, M., Alegre-Cortés, E., Canales-Cortés, S., López de Munain, A., Pérez-Tur, J., Pérez-Castillo, A., Kroemer, G., Fuentes, J. M., & Bravo-San Pedro, J. M. (2023). Changes in Liver Lipidomic Profile in G2019S-LRRK2 Mouse Model of Parkinson’s Disease. Cells, 12(5), 806. https://doi.org/10.3390/cells12050806