
Establishing 20S Proteasome Genetic, Translational and Post-Translational Status from Precious Biological and Patient Samples with Top-Down MS
 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials & Methods
2.1. Reagents
2.2. Cell Lines, Organoids and Crypts
2.2.1. HEK293-EBNA Cells
2.2.2. Caco2 Cells
2.2.3. B lymphoblastoid Cells
2.2.4. Airway Organoids
2.2.5. Intestinal Crypts
2.3. Caco2 Cells Treatment with Different Proinflammatory Cytokines
2.4. Caco2 Cells Treatment with IFNγ
2.5. Purification of c20S and i20S from HEK293-EBNA Cell Lines
2.6. Purification of Proteasome from Caco2 Cells, BLCLs, Organoids and Crypts
2.7. Detailed Top-Down LC-MS Analysis
2.8. Label-Free Quantification in TD-MS using Relative Ionization Yields
2.9. Detailed Bottom-Up LC-MS Analysis
2.10. Proteasome Activity Assay
3. Results
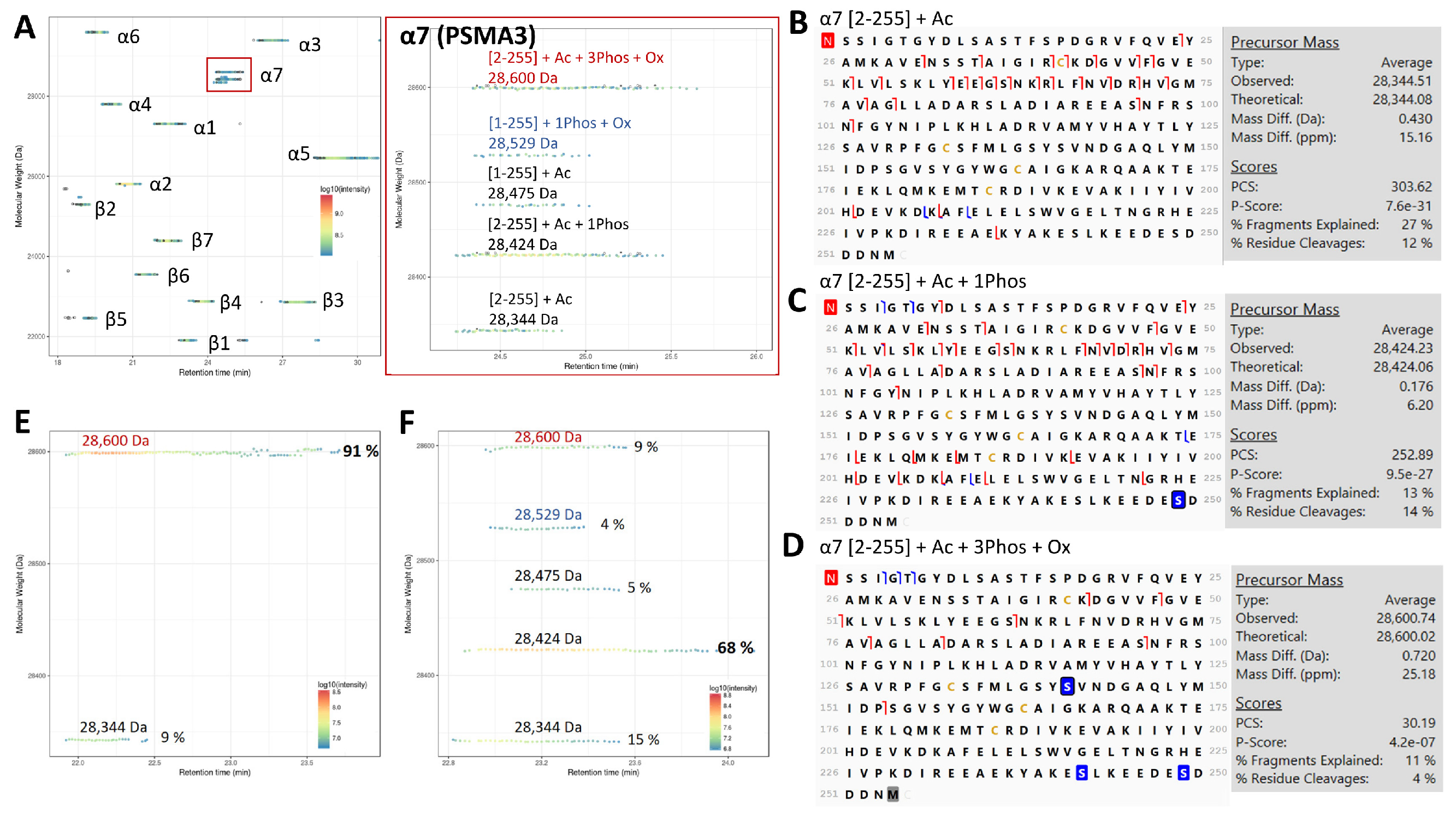
3.1. Establishing 20S Proteoform from Different Biological Materials
3.1.1. Tri-Phosphorylated α7 (PSMA3) in Human 20S Identified by TD-MS
3.1.2. Mutations in 20S Subunits in PRAAS Patients and Their Effect on 20S Assembly
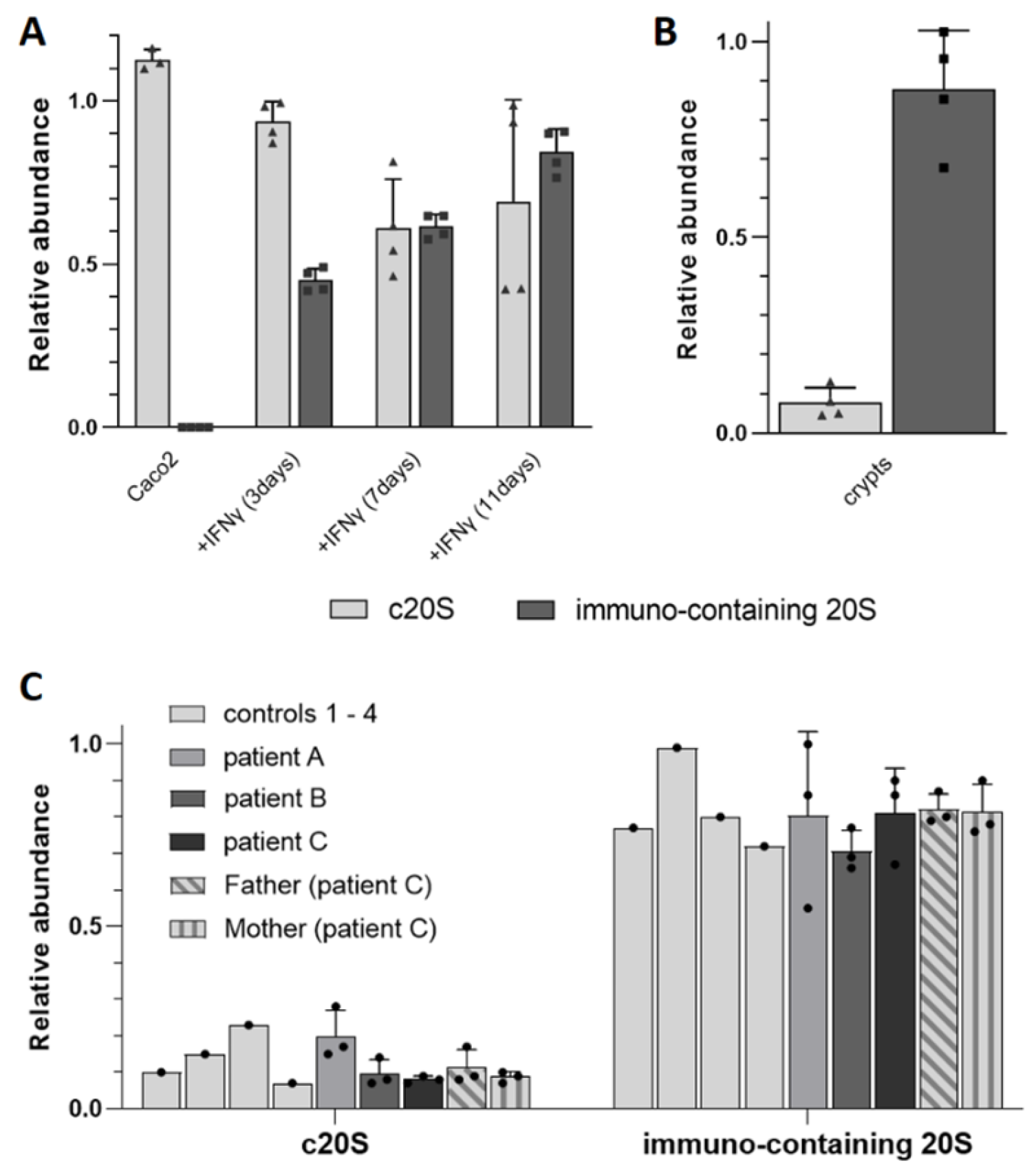
3.1.3. Detection of Immuno Catalytic Subunits in Different Complex Samples by TD-MS
3.2. Relative TD Label-Free Quantification of c20S and i20S in Complex Mixtures
4. Discussion
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kasahara, M.; Flajnik, M.F. Origin and Evolution of the Specialized Forms of Proteasomes Involved in Antigen Presentation. Immunogenetics 2019, 71, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Van den Eynde, B.J. Two Abundant Proteasome Subtypes That Uniquely Process Some Antigens Presented by HLA Class I Molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, M.B.; La Greca, F.; Arastu-Kapur, S.; Caiazza, F.; Cimermancic, P.; Buchholz, T.J.; Anderl, J.L.; Ravalin, M.; Bohn, M.F.; Sali, A.; et al. Immunoproteasome Functions Explained by Divergence in Cleavage Specificity and Regulation. eLife 2017, 6, e27364. [Google Scholar] [CrossRef]
- Raule, M.; Cerruti, F.; Cascio, P. Enhanced Rate of Degradation of Basic Proteins by 26S Immunoproteasomes. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2014, 1843, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Fabre, B.; Lambour, T.; Garrigues, L.; Amalric, F.; Vigneron, N.; Menneteau, T.; Stella, A.; Monsarrat, B.; Van den Eynde, B.; Burlet-Schiltz, O.; et al. Deciphering Preferential Interactions within Supramolecular Protein Complexes: The Proteasome Case. Mol. Syst. Biol. 2015, 11, 771. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.H.; Choi, W.H.; Lee, M.J. Evaluation of Immunoproteasome-Specific Proteolytic Activity Using Fluorogenic Peptide Substrates. Immune Netw. 2022, 22, e28. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Groettrup, M. On the Role of the Immunoproteasome in Protein Homeostasis. Cells 2021, 10, 3216. [Google Scholar] [CrossRef]
- Lesne, J.; Bousquet, M.-P.; Marcoux, J.; Locard-Paulet, M. Top-Down and Intact Protein Mass Spectrometry Data Visualization for Proteoform Analysis Using VisioProt-MS. Bioinform. Biol. Insights 2019, 13, 1177932219868223. [Google Scholar] [CrossRef]
- Živković, D.; Sanchez Dafun, A.; Menneteau, T.; Schahl, A.; Lise, S.; Kervarrec, C.; Toste Rêgo, A.; da Fonseca, P.C.A.; Chavent, M.; Pineau, C.; et al. Proteasome Complexes Experience Profound Structural and Functional Rearrangements throughout Mammalian Spermatogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2116826119. [Google Scholar] [CrossRef]
- Akiyama, K.; Kagawa, S.; Tamura, T.; Shimbara, N.; Takashina, M.; Kristensen, P.; Hendil, K.B.; Tanaka, K.; Ichihara, A. Replacement of Proteasome Subunits X and Y by LMP7 and LMP2 Induced by Interferon-Gamma for Acquirement of the Functional Diversity Responsible for Antigen Processing. FEBS Lett. 1994, 343, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; van den Broek, M.; Schwarz, K.; de Giuli, R.; Diener, P.-A.; Groettrup, M. Immunoproteasomes Largely Replace Constitutive Proteasomes During an Antiviral and Antibacterial Immune Response in the Liver. J. Immunol. 2001, 167, 6859–6868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice Completely Lacking Immunoproteasomes Show Major Changes in Antigen Presentation. Nat. Immunol. 2011, 13, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Moebius, J.; van den Broek, M.; Groettrup, M.; Basler, M. Immunoproteasomes Are Essential for Survival and Expansion of T Cells in Virus-Infected Mice. Eur. J. Immunol. 2010, 40, 3439–3449. [Google Scholar] [CrossRef] [Green Version]
- Abi Habib, J.; De Plaen, E.; Stroobant, V.; Zivkovic, D.; Bousquet, M.-P.; Guillaume, B.; Wahni, K.; Messens, J.; Busse, A.; Vigneron, N.; et al. Efficiency of the Four Proteasome Subtypes to Degrade Ubiquitinated or Oxidized Proteins. Sci. Rep. 2020, 10, 15765. [Google Scholar] [CrossRef]
- Menneteau, T.; Fabre, B.; Garrigues, L.; Stella, A.; Zivkovic, D.; Roux-Dalvai, F.; Mouton-Barbosa, E.; Beau, M.; Renoud, M.-L.; Amalric, F.; et al. Mass Spectrometry-Based Absolute Quantification of 20S Proteasome Status for Controlled Ex-Vivo Expansion of Human Adipose-Derived Mesenchymal Stromal/Stem Cells. Mol. Cell. Proteomics MCP 2019, 18, 744–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J.A. The Immunoproteasome, the 20S Proteasome, and the PA28αβ Proteasome Regulator are Oxidative-Stress-Adaptive Proteolytic Complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehm, A.; Liu, Y.; Sheikh, A.; Marrero, B.; Omoyinmi, E.; Zhou, Q.; Montealegre, G.; Biancotto, A.; Reinhardt, A.; de Jesus, A.A.; et al. Additive Loss-of-Function Proteasome Subunit Mutations in CANDLE/PRAAS Patients Promote Type I IFN Production. J. Clin. Investig. 2015, 125, 4196–4211. [Google Scholar] [CrossRef]
- Davidson, S.; Yu, C.-H.; Steiner, A.; Ebstein, F.; Baker, P.J.; Jarur-Chamy, V.; Hrovat Schaale, K.; Laohamonthonkul, P.; Kong, K.; Calleja, D.J.; et al. Protein Kinase R Is an Innate Immune Sensor of Proteotoxic Stress via Accumulation of Cytoplasmic IL-24. Sci. Immunol. 2022, 7, eabi6763. [Google Scholar] [CrossRef]
- Çetin, G.; Studencka-Turski, M.; Venz, S.; Schormann, E.; Junker, H.; Hammer, E.; Völker, U.; Ebstein, F.; Krüger, E. Immunoproteasomes Control Activation of Innate Immune Signaling and Microglial Function. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Kors, S.; Geijtenbeek, K.; Reits, E.; Schipper-Krom, S. Regulation of Proteasome Activity by (Post-)Transcriptional Mechanisms. Front. Mol. Biosci. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Huang, X.; Chen, M.J. Reversible Phosphorylation of the 26S Proteasome. Protein Cell 2017, 8, 255–272. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.V. Genetics of Proteasome Diseases. Scientifica 2013, 2013, 637629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Cruz, I.; Reynaud, E. Proteasome Subunits Involved in Neurodegenerative Diseases. Arch. Med. Res. 2021, 52, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Coux, O.; Zieba, B.A.; Meiners, S. The Proteasome System in Health and Disease. In Proteostasis and Disease: From Basic Mechanisms to Clinics; Barrio, R., Sutherland, J.D., Rodriguez, M.S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 55–100. ISBN 9783030382667. [Google Scholar]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Xing, C.; DeMartino, G.N.; Mizrachi, D.; Hernandez, M.D.; Sousa, A.B.; Martínez de Villarreal, L.; dos Santos, H.G.; Garg, A. PSMB8 Encoding the Β5i Proteasome Subunit Is Mutated in Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome. Am. J. Hum. Genet. 2010, 87, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Arima, K.; Kinoshita, A.; Mishima, H.; Kanazawa, N.; Kaneko, T.; Mizushima, T.; Ichinose, K.; Nakamura, H.; Tsujino, A.; Kawakami, A.; et al. Proteasome Assembly Defect Due to a Proteasome Subunit Beta Type 8 (PSMB8) Mutation Causes the Autoinflammatory Disorder, Nakajo-Nishimura Syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14914–14919. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A Mutation in the Immunoproteasome Subunit PSMB8 Causes Autoinflammation and Lipodystrophy in Humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.-C.R.; Chen, Y.; et al. Mutations in Proteasome Subunit β Type 8 Cause Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature with Evidence of Genetic and Phenotypic Heterogeneity. Arthritis Rheum. 2012, 64, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Kluk, J.; Rustin, M.; Brogan, P.A.; Omoyinmi, E.; Rowczenio, D.M.; Willcocks, L.C.; Melly, L.; Lachmann, H.J. Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature Syndrome: A Report of a Novel Mutation and Review of the Literature. Br. J. Dermatol. 2014, 170, 215–217. [Google Scholar] [CrossRef] [Green Version]
- Cavalcante, M.P.V.; Brunelli, J.B.; Miranda, C.C.; Novak, G.V.; Malle, L.; Aikawa, N.E.; Jesus, A.A.; Silva, C.A. CANDLE Syndrome: Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature-a Rare Case with a Novel Mutation. Eur. J. Pediatr. 2016, 175, 735–740. [Google Scholar] [CrossRef]
- Tüfekçi, Ö.; Bengoa, S.; Karapinar, T.H.; Ataseven, E.B.; Irken, G.; Ören, H. CANDLE Syndrome: A Recently Described Autoinflammatory Syndrome. J. Pediatr. Hematol. Oncol. 2015, 37, 296–299. [Google Scholar] [CrossRef]
- Kanazawa, N.; Hemmi, H.; Kinjo, N.; Ohnishi, H.; Hamazaki, J.; Mishima, H.; Kinoshita, A.; Mizushima, T.; Hamada, S.; Hamada, K.; et al. Heterozygous Missense Variant of the Proteasome Subunit β-Type 9 Causes Neonatal-Onset Autoinflammation and Immunodeficiency. Nat. Commun. 2021, 12, 6819. [Google Scholar] [CrossRef] [PubMed]
- Sarrabay, G.; Méchin, D.; Salhi, A.; Boursier, G.; Rittore, C.; Crow, Y.; Rice, G.; Tran, T.-A.; Cezar, R.; Duffy, D.; et al. PSMB10, the Last Immunoproteasome Gene Missing for PRAAS. J. Allergy Clin. Immunol. 2020, 145, 1015–1017.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, D.; Schonenberg-Meinema, D.; Ebstein, F.; Papendorf, J.J.; Baars, P.A.; van Leeuwen, E.M.M.; Jansen, M.H.; Lankester, A.C.; van der Burg, M.; Florquin, S.; et al. Hematopoietic Stem Cell Transplantation in a Patient with Proteasome-Associated Autoinflammatory Syndrome (PRAAS). J. Allergy Clin. Immunol. 2022, 149, 1120–1127.e8. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.A.; Brehm, A.; VanTries, R.; Pillet, P.; Parentelli, A.-S.; Montealegre Sanchez, G.A.; Deng, Z.; Paut, I.K.; Goldbach-Mansky, R.; Krüger, E. Novel Proteasome Assembly Chaperone Mutations in PSMG2/PAC2, Cause the Autoinflammatory Interferonopathy, CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019, 143, 1939–1943.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; de Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef] [Green Version]
- Küry, S.; Besnard, T.; Ebstein, F.; Khan, T.N.; Gambin, T.; Douglas, J.; Bacino, C.A.; Craigen, W.J.; Sanders, S.J.; Lehmann, A.; et al. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 100, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Isidor, B.; Ebstein, F.; Hurst, A.; Vincent, M.; Bader, I.; Rudy, N.L.; Cogne, B.; Mayr, J.; Brehm, A.; Bupp, C.; et al. Stankiewicz-Isidor Syndrome: Expanding the Clinical and Molecular Phenotype. Genet. Med. Off. J. Am. Coll. Med. Genet. 2022, 24, 179–191. [Google Scholar] [CrossRef]
- Kröll-Hermi, A.; Ebstein, F.; Stoetzel, C.; Geoffroy, V.; Schaefer, E.; Scheidecker, S.; Bär, S.; Takamiya, M.; Kawakami, K.; Zieba, B.A.; et al. Proteasome Subunit PSMC3 Variants Cause Neurosensory Syndrome Combining Deafness and Cataract Due to Proteotoxic Stress. EMBO Mol. Med. 2020, 12, e11861. [Google Scholar] [CrossRef]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-Mediated 26S Proteasome Inhibition Causes Cell-Cycle Arrest and Induces Apoptosis in CD-30+ Anaplastic Large Cell Lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.B.; Chanan-Khan, A.A.; et al. Potent Activity of Carfilzomib, a Novel, Irreversible Inhibitor of the Ubiquitin-Proteasome Pathway, against Preclinical Models of Multiple Myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Groettrup, M. Recent Insights How Combined Inhibition of Immuno/Proteasome Subunits Enables Therapeutic Efficacy. Genes Immun. 2020, 21, 273–287. [Google Scholar] [CrossRef]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome Inhibition Prevents Chronic Antibody-Mediated Allograft Rejection in Renal Transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koerner, J.; Brunner, T.; Groettrup, M. Inhibition and Deficiency of the Immunoproteasome Subunit LMP7 Suppress the Development and Progression of Colorectal Carcinoma in Mice. Oncotarget 2017, 8, 50873–50888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basler, M.; Christ, M.; Goebel, H.; Groettrup, M. Immunoproteasome Upregulation Is Not Required to Control Protein Homeostasis during Viral Infection. J. Immunol. 2021, 206, 1697–1708. [Google Scholar] [CrossRef]
- Wani, P.S.; Suppahia, A.; Capalla, X.; Ondracek, A.; Roelofs, J. Phosphorylation of the C-Terminal Tail of Proteasome Subunit A7 Is Required for Binding of the Proteasome Quality Control Factor Ecm29. Sci. Rep. 2016, 6, 27873. [Google Scholar] [CrossRef] [Green Version]
- Froment, C.; Uttenweiler-Joseph, S.; Bousquet-Dubouch, M.-P.; Matondo, M.; Borges, J.-P.; Esmenjaud, C.; Lacroix, C.; Monsarrat, B.; Burlet-Schiltz, O. A Quantitative Proteomic Approach Using Two-Dimensional Gel Electrophoresis and Isotope-Coded Affinity Tag Labeling for Studying Human 20S Proteasome Heterogeneity. Proteomics 2005, 5, 2351–2363. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Delobel, J.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Subcellular Distribution and Dynamics of Active Proteasome Complexes Unraveled by a Workflow Combining in Vivo Complex Cross-Linking and Quantitative Proteomics. Mol. Cell. Proteomics MCP 2013, 12, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down Proteomics. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Durbin, K.R.; Fornelli, L.; Fellers, R.T.; Doubleday, P.F.; Narita, M.; Kelleher, N.L. Quantitation and Identification of Thousands of Human Proteoforms below 30 KDa. J. Proteome Res. 2016, 15, 976–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersch, M.; Hackl, M.W.; Dubiella, C.; Dobrinevski, A.; Groll, M.; Sieber, S.A. A Mass Spectrometry Platform for a Streamlined Investigation of Proteasome Integrity, Posttranslational Modifications, and Inhibitor Binding. Chem. Biol. 2015, 22, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupp-Sutton, K.A.; Wu, S. High-Throughput Quantitative Top-down Proteomics. Mol. Omics 2020, 16, 91–99. [Google Scholar] [CrossRef]
- Collier, T.S.; Sarkar, P.; Rao, B.; Muddiman, D.C. Quantitative Top-down Proteomics of SILAC Labeled Human Embryonic Stem Cells. J. Am. Soc. Mass Spectrom. 2010, 21, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Winkels, K.; Koudelka, T.; Tholey, A. Quantitative Top-Down Proteomics by Isobaric Labeling with Thiol-Directed Tandem Mass Tags. J. Proteome Res. 2021, 20, 4495–4506. [Google Scholar] [CrossRef]
- Yu, D.; Wang, Z.; Cupp-Sutton, K.A.; Guo, Y.; Kou, Q.; Smith, K.; Liu, X.; Wu, S. Quantitative Top-Down Proteomics in Complex Samples Using Protein-Level Tandem Mass Tag Labeling. J. Am. Soc. Mass Spectrom. 2021, 32, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, P.; Wang, D. Quantitation of Intact Proteins in Human Plasma Using Top-Down Parallel Reaction Monitoring-MS. Anal. Chem. 2018, 90, 10650–10653. [Google Scholar] [CrossRef]
- Ntai, I.; Toby, T.K.; LeDuc, R.D.; Kelleher, N.L. A Method for Label-Free, Differential Top-Down Proteomics. Methods Mol. Biol. 2016, 1410, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Geis-Asteggiante, L.; Ostrand-Rosenberg, S.; Fenselau, C.; Edwards, N.J. Evaluation of Spectral Counting for Relative Quantitation of Proteoforms in Top-Down Proteomics. Anal. Chem. 2016, 88, 10900–10907. [Google Scholar] [CrossRef] [PubMed]
- Iakobachvili, N.; Leon-Icaza, S.A.; Knoops, K.; Sachs, N.; Mazères, S.; Simeone, R.; Peixoto, A.; Bernard, C.; Murris-Espin, M.; Mazières, J.; et al. Mycobacteria–Host Interactions in Human Bronchiolar Airway Organoids. Mol. Microbiol. 2022, 117, 682–692. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Bousquet-Dubouch, M.-P.; Baudelet, E.; Guérin, F.; Matondo, M.; Uttenweiler-Joseph, S.; Burlet-Schiltz, O.; Monsarrat, B. Affinity Purification Strategy to Capture Human Endogenous Proteasome Complexes Diversity and to Identify Proteasome-Interacting Proteins*S. Mol. Cell. Proteomics 2009, 8, 1150–1164. [Google Scholar] [CrossRef] [Green Version]
- Locard-Paulet, M.; Parra, J.; Albigot, R.; Mouton-Barbosa, E.; Bardi, L.; Burlet-Schiltz, O.; Marcoux, J. VisioProt-MS: Interactive 2D Maps from Intact Protein Mass Spectrometry. Bioinformatics 2019, 35, 679–681. [Google Scholar] [CrossRef]
- Marty, M.T.; Baldwin, A.J.; Marklund, E.G.; Hochberg, G.K.A.; Benesch, J.L.P.; Robinson, C.V. Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles. Anal. Chem. 2015, 87, 4370–4376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouyssié, D.; Hesse, A.-M.; Mouton-Barbosa, E.; Rompais, M.; Macron, C.; Carapito, C.; Gonzalez de Peredo, A.; Couté, Y.; Dupierris, V.; Burel, A.; et al. Proline: An Efficient and User-Friendly Software Suite for Large-Scale Proteomics. Bioinformatics 2020, 36, 3148–3155. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Stratford, F.L.L.; Broadfoot, K.I.; Mason, G.G.F.; Rivett, A.J. Phosphorylation of 20S Proteasome Alpha Subunit C8 (Alpha7) Stabilizes the 26S Proteasome and Plays a Role in the Regulation of Proteasome Complexes by Gamma-Interferon. Biochem. J. 2004, 378, 177–184. [Google Scholar] [CrossRef]
- Schmidt, F.; Dahlmann, B.; Hustoft, H.K.; Koehler, C.J.; Strozynski, M.; Kloß, A.; Zimny-Arndt, U.; Jungblut, P.R.; Thiede, B. Quantitative Proteome Analysis of the 20S Proteasome of Apoptotic Jurkat T Cells. Amino Acids 2011, 41, 351–361. [Google Scholar] [CrossRef]
- Castaño, J.G.; Mahillo, E.; Arizti, P.; Arribas, J. Phosphorylation of C8 and C9 Subunits of the Multicatalytic Proteinase by Casein Kinase II and Identification of the C8 Phosphorylation Sites by Direct Mutagenesis. Biochemistry 1996, 35, 3782–3789. [Google Scholar] [CrossRef]
- Mason, G.G.F.; Murray, R.Z.; Pappin, D.; Rivett, A.J. Phosphorylation of ATPase Subunits of the 26S Proteasome. FEBS Lett. 1998, 430, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, C.-F.; Baker, P.R.; Chen, P.; Kaiser, P.; Huang, L. Mass Spectrometric Characterization of the Affinity-Purified Human 26S Proteasome Complex. Biochemistry 2007, 46, 3553–3565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.R.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and Structure-Based Prediction of Eukaryotic Protein Phosphorylation Sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of Post-Translational Glycosylation and Phosphorylation of Proteins from the Amino Acid Sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Claverol, S.; Burlet-Schiltz, O.; Girbal-Neuhauser, E.; Gairin, J.E.; Monsarrat, B. Mapping and Structural Dissection of Human 20 S Proteasome Using Proteomic Approaches. Mol. Cell. Proteomics 2002, 1, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.C.; Marques, A.J.; London, M.K.; Dohmen, R.J. Role of C-Terminal Extensions of Subunits Β2 and Β7 in Assembly and Activity of Eukaryotic Proteasomes*. J. Biol. Chem. 2004, 279, 14323–14330. [Google Scholar] [CrossRef] [Green Version]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.E.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-Mediated Degradation of IκBα and Processing of P105 in Crohn Disease and Ulcerative Colitis. J. Clin. Investig. 2006, 116, 3195–3203. [Google Scholar] [CrossRef] [Green Version]
- Keller, I.E.; Vosyka, O.; Takenaka, S.; Kloß, A.; Dahlmann, B.; Willems, L.I.; Verdoes, M.; Overkleeft, H.S.; Marcos, E.; Adnot, S.; et al. Regulation of Immunoproteasome Function in the Lung. Sci. Rep. 2015, 5, 10230. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. J. Immunol. Res. 2015, 2015, e541984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Kammerl, I.; Eickelberg, O.; Schamberger, A.; Meiners, S. Role of the Immunoproteasome during of Bronchial Epithelial Cell Differentiation. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef]
- Carel, C.; Marcoux, J.; Réat, V.; Parra, J.; Latgé, G.; Laval, F.; Demange, P.; Burlet-Schiltz, O.; Milon, A.; Daffé, M.; et al. Identification of Specific Posttranslational O-Mycoloylations Mediating Protein Targeting to the Mycomembrane. Proc. Natl. Acad. Sci. USA 2017, 114, 4231–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, J.; Marcoux, J.; Poncin, I.; Canaan, S.; Herrmann, J.L.; Nigou, J.; Burlet-Schiltz, O.; Rivière, M. Scrutiny of Mycobacterium Tuberculosis 19 KDa Antigen Proteoforms Provides New Insights in the Lipoglycoprotein Biogenesis Paradigm. Sci. Rep. 2017, 7, 43682. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Su, K.; Yang, X.; Bowe, D.B.; Paterson, A.J.; Kudlow, J.E. O-GlcNAc Modification Is an Endogenous Inhibitor of the Proteasome. Cell 2003, 115, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, S.; Viollet, B.; Zou, M.-H. Regulation of the Proteasome by AMPK in Endothelial Cells: The Role of O-GlcNAc Transferase (OGT). PLOS ONE 2012, 7, e36717. [Google Scholar] [CrossRef]
- Pecori, F.; Kondo, N.; Ogura, C.; Miura, T.; Kume, M.; Minamijima, Y.; Yamamoto, K.; Nishihara, S. Site-Specific O-GlcNAcylation of Psme3 Maintains Mouse Stem Cell Pluripotency by Impairing P-Body Homeostasis. Cell Rep. 2021, 36, 109361. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.; Silva, G.M.; Netto, L.E.S. 20 S Proteasome from Saccharomyces Cerevisiae Is Responsive to Redox Modifications and Is S-Glutathionylated. J. Biol. Chem. 2003, 278, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Cho-Park, P.F.; Steller, H. Proteasome Regulation by ADP-Ribosylation. Cell 2013, 153, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Tsimokha, A.S.; Artamonova, T.O.; Diakonov, E.E.; Khodorkovskii, M.A.; Tomilin, A.N. Post-Translational Modifications of Extracellular Proteasome. Molecules 2020, 25, 3504. [Google Scholar] [CrossRef]
- Kimura, A.; Kato, Y.; Hirano, H. N-Myristoylation of the Rpt2 Subunit Regulates Intracellular Localization of the Yeast 26S Proteasome. Biochemistry 2012, 51, 8856–8866. [Google Scholar] [CrossRef]
- Bai, M.; Zhao, X.; Sahara, K.; Ohte, Y.; Hirano, Y.; Kaneko, T.; Yashiroda, H.; Murata, S. Assembly Mechanisms of Specialized Core Particles of the Proteasome. Biomolecules 2014, 4, 662–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, Y.; Kaneko, T.; Okamoto, K.; Bai, M.; Yashiroda, H.; Furuyama, K.; Kato, K.; Tanaka, K.; Murata, S. Dissecting β-Ring Assembly Pathway of the Mammalian 20S Proteasome. EMBO J. 2008, 27, 2204–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.-L.; Dong, C.; Andreev, V.; Arcos-Burgos, M.; Licinio, J. Prediction of Susceptibility to Major Depression by a Model of Interactions of Multiple Functional Genetic Variants and Environmental Factors. Mol. Psychiatry 2012, 17, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Huguet, R.; Mullen, C.; Srzentić, K.; Greer, J.B.; Fellers, R.T.; Zabrouskov, V.; Syka, J.E.P.; Kelleher, N.L.; Fornelli, L. Proton Transfer Charge Reduction Enables High-Throughput Top-Down Analysis of Large Proteoforms. Anal. Chem. 2019, 91, 15732–15739. [Google Scholar] [CrossRef] [PubMed]
- Kafader, J.O.; Durbin, K.R.; Melani, R.D.; Des Soye, B.J.; Schachner, L.F.; Senko, M.W.; Compton, P.D.; Kelleher, N.L. Individual Ion Mass Spectrometry Enhances the Sensitivity and Sequence Coverage of Top-Down Mass Spectrometry. J. Proteome Res. 2020, 19, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Blevins, M.S.; Juetten, K.J.; James, V.K.; Butalewicz, J.P.; Escobar, E.E.; Lanzillotti, M.B.; Sanders, J.D.; Fort, K.L.; Brodbelt, J.S. Nanohydrophobic Interaction Chromatography Coupled to Ultraviolet Photodissociation Mass Spectrometry for the Analysis of Intact Proteins in Low Charge States. J. Proteome Res. 2022, 21, 2493–2503. [Google Scholar] [CrossRef]
- Greisch, J.-F.; den Boer, M.A.; Lai, S.-H.; Gallagher, K.; Bondt, A.; Commandeur, J.; Heck, A.J.R. Extending Native Top-Down Electron Capture Dissociation to MDa Immunoglobulin Complexes Provides Useful Sequence Tags Covering Their Critical Variable Complementarity-Determining Regions. Anal. Chem. 2021, 93, 16068–16075. [Google Scholar] [CrossRef]
- Harvey, S.R.; Ben-Nissan, G.; Sharon, M.; Wysocki, V.H. Surface-Induced Dissociation for Protein Complex Characterization. Methods Mol. Biol. Clifton NJ 2022, 2500, 211–237. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Years) | Sex | Medical Status |
|---|---|---|
| 66 | M | Low-grade pseudoperitoneal myxoma of appendicular origin |
| 59 | M | Suspicion of colorectal cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dafun, A.S.; Živković, D.; Leon-Icaza, S.A.; Möller, S.; Froment, C.; Bonnet, D.; de Jesus, A.A.; Alric, L.; Quaranta-Nicaise, M.; Ferrand, A.; et al. Establishing 20S Proteasome Genetic, Translational and Post-Translational Status from Precious Biological and Patient Samples with Top-Down MS. Cells 2023, 12, 844. https://doi.org/10.3390/cells12060844
Dafun AS, Živković D, Leon-Icaza SA, Möller S, Froment C, Bonnet D, de Jesus AA, Alric L, Quaranta-Nicaise M, Ferrand A, et al. Establishing 20S Proteasome Genetic, Translational and Post-Translational Status from Precious Biological and Patient Samples with Top-Down MS. Cells. 2023; 12(6):844. https://doi.org/10.3390/cells12060844
Chicago/Turabian StyleDafun, Angelique Sanchez, Dušan Živković, Stephen Adonai Leon-Icaza, Sophie Möller, Carine Froment, Delphine Bonnet, Adriana Almeida de Jesus, Laurent Alric, Muriel Quaranta-Nicaise, Audrey Ferrand, and et al. 2023. "Establishing 20S Proteasome Genetic, Translational and Post-Translational Status from Precious Biological and Patient Samples with Top-Down MS" Cells 12, no. 6: 844. https://doi.org/10.3390/cells12060844