
Mechanistic Insights into the Role of OPN in Mediating Brain Damage via Triggering Lysosomal Damage in Microglia/Macrophage
Abstract
:
1. Introduction
2. Methods
2.1. Ethical Statement
2.2. Animals and Treatment
2.3. Sample Preparation for Tandem Mass Tag (TMT) Analysis
2.4. Protein–Protein Interaction (PPI) Network
2.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analyses
2.6. Lentiviral and Plasmid Transfection
2.7. Oxygen–Glucose Deprivation (OGD)
2.8. Lyso-Tracker Red and Mito-Tracker Red CMXRos Staining
2.9. Analysis of Lysosome Membrane Permeability
2.10. Infarct Volume Measurement
2.11. Lateral Cerebral Ventricle Injections
2.12. Immunofluorescence Staining
2.13. Terminal Deoxynucleotidyltransferase-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Assay
2.14. Co-Immunoprecipitation (IP) Assay
2.15. Neurobehavioral Evaluation
2.15.1. Geotaxis Reflex
2.15.2. Grip Test
2.15.3. T-Maze
2.16. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.17. Western Blot Analysis
2.18. Statistical Analysis
3. Results
3.1. Up-Regulated OPN Colocalized with LAMP1 and GAL-3 in Microglia/Macrophages after HI Insult
3.2. HI Insult Led to Impairment of Lysosomal Function and Autophagic Flux
3.3. OPN Deficiency Reduced HI-Induced Lysosomes Damage and Autophagosome Accumulation, Associating with Improving Behavior Deficit
3.4. OPN Deficiency Reduced CTSB Release into Cytoplasm and NLRP3 Inflammasome Activation after HI Insult
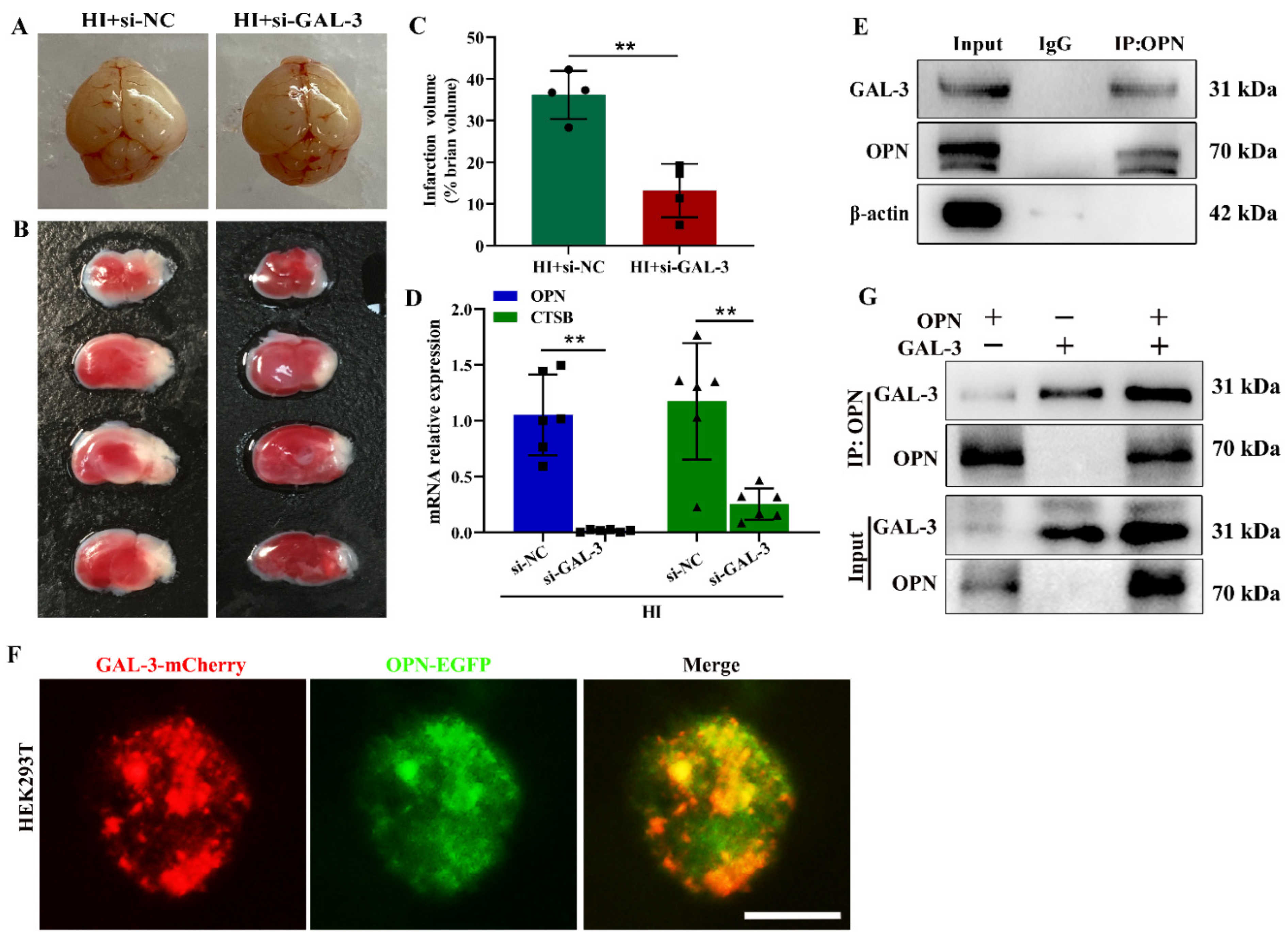
3.5. OPN Interacts with GAL-3 to Induced Lysosomal Damage following HI Exposure
3.6. Cleaved OPN Promoted Lysosomal Damage by Interacting with GAL-3
3.7. Role of CD44 in OPN-Induced Lysosomal Damage following HI Insult
3.8. Secreted OPN Stimulated Pro-Inflammatory Cytokines and CTSB Release through Binding with Cell Surface Receptors CD44
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenin |
| AO | Acridine orange |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| co-IP | Co-immunoprecipitation |
| CQ | Chloroquine |
| CTSB | Cathepsin B |
| DAPI | 4′,6′-Diamidino-2-phenylindole dihydrochloride hydrate |
| DEPs | Differentially expressed proteins |
| GAL-3 | Galectin-3 |
| GFAP | Glial fibrillary acidic protein |
| HI | Hypoxia–ischemia |
| Iba-1 | Ionized calcium-binding adapter molecule 1 |
| IL-1β | Interleukin-1β |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| MMP | Matrix metalloproteinases |
| NC | Negative control |
| NLRP3 | NOD-like receptor thermal protein domain associated protein 3 |
| NeuN | Neuronal specific nuclear protein |
| OGD | Oxygen–glucose deprivation |
| OPN | Osteopontin |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| rmOPN | Recombinant mouse OPN |
| TUNEL | Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling |
| TTC | 2,3,5-Triphenyltetrazolium chloride monohydrate |
References
- Bellahcène, A.; Castronovo, V.; Ogbureke, K.U.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 2008, 8, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.P.; Jander, S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.L.; Reeves, T.M.; Phillips, L.L. Phillips. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp. Neurol. 2014, 261, 757–771. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Qiu, Y.; Wight, A.E.; Kim, H.J.; Cantor, H. Definition of a mouse microglial subset that regulates neuronal development and proinflammatory responses in the brain. Proc. Natl. Acad. Sci. USA 2022, 119, e2116241119. [Google Scholar] [CrossRef]
- Yim, A.; Smith, C.; Brown, A.M. Osteopontin/secreted phosphoprotein-1 harnesses glial-, immune-, and neuronal cell ligand-receptor interactions to sense and regulate acute and chronic neuroinflammation. Immunol. Rev. 2022, 311, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Xin, D.; Li, T.; Chu, X.; Ke, H.; Liu, D.; Wang, Z. MSCs-extracellular vesicles attenuated neuroinflammation, synapse damage and microglial phagocytosis after hypoxia-ischemia injury by preventing osteopontin expression. Pharmacol. Res. 2021, 164, 105322. [Google Scholar] [CrossRef]
- Li, T.-T.; Xin, D.-Q.; Ke, H.-F.; Chu, X.-L.; Zhao, Y.-J.; Yue, S.-W.; Liu, D.-X.; Wang, Z. L-Cysteine attenuates osteopontin-mediated neuroinflammation following hypoxia-ischemia insult in neonatal mice by inducing S-sulfhydration of Stat3. Acta Pharmacol. Sin. 2021, 43, 1658–1669. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yao, Q.; Hu, T.; Cai, Z.; Xie, Q.; Zhao, J.; Yuan, Y.; Ni, J.; Wu, Q.Q. Cathepsin B deteriorates diabetic cardiomyopathy induced by streptozotocin via promoting NLRP3-mediated pyroptosis. Mol. Ther. Nucleic Acids 2022, 30, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, Y.; Chen, H.; Tao, M.; Zhou, X.; Li, J.; Ma, X.; Wang, Y.; Liu, N. Blockade of autophagy prevents the progression of hyperuricemic nephropathy through inhibiting NLRP3 inflammasome-mediated pyroptosis. Front. Immunol. 2022, 13, 858494. [Google Scholar] [CrossRef]
- Stone, B.S.; Zhang, J.; Mack, D.W.; Mori, S.; Martin, L.J.; Northington, F.J. Delayed neural network degeneration after neonatal hypoxia-ischemia. Ann. Neurol. 2008, 64, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Sun, G.; Chen, B.; Xu, L.; Ye, Y.; He, J.; Bao, Z.; Zhao, P.; Miao, Z.; Zhao, L.; et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res. 2021, 174, 105933. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, D.; Ren, Y.; Zhang, Z.; Guo, X.; Ma, M.; Xue, Z.; Lv, J.; Liu, H.; Xi, Q.; et al. Mir223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy 2019, 15, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.-P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altern. Anim. Exp. 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ding, K.; Ji, J.; Parajuli, H.; Aasen, S.N.; Espedal, H.; Huang, B.; Chen, A.; Wang, J.; Li, X.; et al. Trifluoperazine prolongs the survival of experimental brain metastases by STAT3-dependent lysosomal membrane permeabilization. Am. J. Cancer Res. 2020, 10, 545–563. [Google Scholar]
- Bai, X.; Liu, S.; Yuan, L.; Xie, Y.; Li, T.; Wang, L.; Wang, X.; Zhang, T.; Qin, S.; Song, G.; et al. Hydrogen-rich saline mediates neuroprotection through the regulation of endoplasmic reticulum stress and autophagy under hypoxia-ischemia neonatal brain injury in mice. Brain Res. 2016, 1646, 410–417. [Google Scholar] [CrossRef]
- Kim, J.Y.; Grunke, S.D.; Levites, Y.; Golde, T.E.; Jankowsky, J.L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp. JoVE 2014, 91, 51863. [Google Scholar]
- Li, T.; Chu, X.; Xin, D.; Ke, H.; Wang, S.; Liu, D.; Chen, W.; Wang, Z. H2S prevents peripheral immune cell invasion, increasing [Ca2+]i and excessive phagocytosis following hypoxia-ischemia injury in neonatal mice. Biomed. Pharmacother. 2021, 135, 111207. [Google Scholar] [CrossRef]
- Rotshenker, S. Galectin-3 (MAC-2) controls phagocytosis and macropinocytosis through intracellular and extracellular mechanisms. Front. Cell. Neurosci. 2022, 16, 949079. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zhao, J.; Zhang, Y.; Liu, Y.; Ma, C.; Yi, F.; Zheng, Y.; Zhang, L.; Chen, T.; Liu, H.; et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy 2022, 18, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Siew, J.J.; Chen, H.M.; Chen, H.Y.; Chen, H.L.; Chen, C.M.; Soong, B.W.; Wu, Y.R.; Chang, C.P.; Chan, Y.C.; Lin, C.H.; et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 2019, 10, 3473. [Google Scholar] [CrossRef] [Green Version]
- Xin, D.; Chu, X.; Bai, X.; Ma, W.; Yuan, H.; Qiu, J.; Liu, C.; Li, T.; Zhou, X.; Chen, W.; et al. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behav. Immun. 2018, 73, 222–234. [Google Scholar] [CrossRef]
- Ni, J.; Wu, Z.; Peterts, C.; Yamamoto, K.; Qing, H.; Nakanishi, H. The critical role of proteolytic relay through cathepsins B and E in the phenotypic change of microglia/macrophage. J. Neurosci. 2015, 35, 12488–12501. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Claude-Taupin, A.; Gu, Y.; Choi, S.W.; Peters, R.; Bissa, B.; Mudd, M.H.; Allers, L.; Pallikkuth, S.; Lidke, K.A.; et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 2020, 52, 69–87.e8. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Zouein, F.A.; Tian, Y.; Padmanabhan Iyer, R.; de Castro Brás, L.E. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can. J. Physiol. Pharmacol. 2015, 93, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Peraramelli, S.; Zhou, Q.; Zhou, Q.; Wanko, B.; Zhao, L.; Nishimura, T.; Leung, T.H.; Mizuno, S.; Ito, M.; Myles, T.; et al. Thrombin cleavage of osteopontin initiates osteopontin’s tumor-promoting activity. J. Thromb. Haemost. 2022, 20, 1256–1270. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015, 1623, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhan, Y.; Xu, L.; Feuerstein, G.Z.; Wang, W. Use of suppression subtractive hybridization for differential gene expression in stroke: Discovery of CD44 gene expression and localization in permanent focal stroke in rats. Stroke 2001, 32, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.S.; Choi, J.S.; Shin, Y.J.; Kim, H.Y.; Cha, J.H.; Lee, J.Y.; Chun, M.H.; Lee, M.Y. Differential regulation of osteopontin receptors, CD44 and the alpha(v) and beta(3) integrin subunits, in the rat hippocampus following transient forebrain ischemia. Brain Res. 2008, 1228, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Troncoso, M.; Bannoud, N.; Carvelli, L.; Asensio, J.; Seltzer, A.; Sosa, M.A. Hypoxia-ischemia alters distribution of lysosomal proteins in rat cortex and hippocampus. Biol. Open 2018, 7, bio036723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Blázquez, P.; Pistorio, V.; Fernández-Fernández, M.; Moles, A. The multifaceted role of cathepsins in liver disease. J. Hepatol. 2021, 75, 1192–1202. [Google Scholar] [CrossRef]
- Bai, H.; Yang, B.; Yu, W.; Xiao, Y.; Yu, D.; Zhang, Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 2018, 362, 180–187. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangère, V.; Chalmin, F.; Chevriaux, A.; Végran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef]
- Tang, T.T.; Lv, L.L.; Pan, M.M.; Wen, Y.; Wang, B.; Li, Z.L.; Wu, M.; Wang, F.M.; Crowley, S.D.; Liu, B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wei, M.; Fan, J.; Yan, W.; Zha, X.; Song, H.; Wan, R.; Yin, Y.; Wang, W. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 2021, 17, 1519–1542. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Yoo, H.-I.; Kim, E.-G.; Lee, E.-J.; Hong, S.-Y.; Yoon, C.-S.; Hong, M.-J.; Park, S.-J.; Woo, R.-S.; Baik, T.-K.; Song, D.Y. Neuroanatomical distribution of galectin-3 in the adult rat brain. J. Mol. Histol. 2017, 48, 133–146. [Google Scholar] [CrossRef]
- Puigdellívol, M.; Allendorf, D.H.; Brown, G.C. Sialylation and galectin-3 in microglia-mediated neuroinflammation and neurodegeneration. Front. Cell. Neurosci. 2020, 14, 162. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zheng, Y.; Xu, D.; Sun, Z.; Yang, H.; Yin, Q. Galectin-3: A key player in microglia-mediated neuroinflammation and Alzheimer’s disease. Cell Biosci. 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hébert, M.; Swarup, V.; Beaulieu, J.M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 2012, 32, 10383–10395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, K.; Ma, Y.; Li, S.; Xu, Y. Serum Galectin-3 as a potential predictive biomarker is associated with poststroke cognitive impairment. Oxidative Med. Cell. Longev. 2021, 2021, 5827812. [Google Scholar] [CrossRef] [PubMed]
- Sävman, K.; Heyes, M.P.; Svedin, P.; Karlsson, A. Microglia/macrophage-derived inflammatory mediators galectin-3 and quinolinic acid are elevated in cerebrospinal fluid from newborn infants after birth asphyxia. Transl. Stroke Res. 2013, 4, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Diao, S.; Qiu, H.; Gao, R.; Wang, M.; Chen, Q.; Xiao, M.; Li, Z.; Chen, C. Galectin-3 administration drives remyelination after hypoxic-ischemic induced perinatal white matter injury. Front. Cell. Neurosci. 2022, 16, 976002. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doverhag, C.; Hedtjärn, M.; Poirier, F.; Mallard, C.; Hagberg, H.; Karlsson, A.; Sävman, K. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2010, 38, 36–46. [Google Scholar] [CrossRef]
- Gao, Y.A.; Agnihotri, R.; Vary, C.P.H.; Liaw, L. Expression and characterization of recombinant osteopontin peptides representing matrix metalloproteinase proteolytic fragments. Matrix Biol. 2004, 23, 457–466. [Google Scholar] [CrossRef]
- Kon, S.; Nakayama, Y.; Matsumoto, N.; Ito, K.; Kanayama, M.; Kimura, C.; Kouro, H.; Ashitomi, D.; Matsuda, T.; Uede, T. A novel cryptic binding motif, LRSKSRSFQVSDEQY, in the C-terminal fragment of MMP-3/7-cleaved osteopontin as a novel ligand for α9β1 integrin is involved in the anti-type II collagen antibody-induced arthritis. PLoS ONE 2014, 9, e116210. [Google Scholar] [CrossRef] [Green Version]
- Hamias, R.; Rudich, A.; Greenberg, G.; Szendro, G.; Wolak, T. Angiotensin 1-7, but not the thrombin-cleaved osteopontin C-terminal fragment, attenuates osteopontin-mediated macrophage-induced endothelial-cell inflammation. Inflamm. Res. 2018, 67, 265–275. [Google Scholar] [CrossRef]
- Rangaswami, H.; Bulbule, A.; Kundu, G.C. Osteopontin: Role in cell signaling and cancer progression. Trends Cell Biol. 2006, 16, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sarkar-Dutta, M.; McRae, S.; Ramachandran, A.; Kumar, B.; Waris, G. Osteopontin regulates Hepatitis C virus (HCV) replication and assembly by interacting with HCV proteins and lipid droplets and by binding to receptors αVβ3 and CD44. J. Virol. 2018, 92, e02116–e02117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, S.; Raja, R.; Thorat, D.; Soundararajan, G.; Patil, T.V.; Kundu, G.C. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via α9β1 integrin. Oncogene 2014, 33, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Sottnik, J.L.; Dancik, G.M.; Sahu, D.; Hansel, D.E.; Theodorescu, D.; Schwartz, M.A. An osteopontin/CD44 axis in RhoGDI2-mediated metastasis suppression. Cancer Cell 2016, 30, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Catalog | Company | Dilution | |
|---|---|---|---|---|
| Number | IF | WB | ||
| Mouse β-actin antibody | TA-09 | Zhongshan Golden Bridge Biotechnology | 1:1000 | |
| Rabbit OPN antibody | 22952-1-AP | Proteintech | 1:200 | 1:1000 |
| Mouse GAL-3 antibody | sc-32790 | Santa Cruz | 1:200 | 1:1000 |
| Rabbit NLPR3 antibody | ab263899 | Abcam | 1:1000 | |
| Rabbit CTSB antibody | 12216-1-AP | Proteintech | 1:200 | 1:1000 |
| Rabbit LAMP1 antibody | ab24170 | Abcam | 1:1000 | |
| Rabbit IL-1β antibody | sc-7884 | Santa Cruz | 1:1000 | |
| Mouse NeuN antibody | ab104224 | Abcam | 1:200 | |
| Mouse Iba-1 antibody | 66827-1-Ig | Proteintech | 1:200 | |
| Mouse GFAP antibody | 60190–1-Ig | Proteintech | 1:200 | |
| Rabbit LC3B antibody | #2775 | Cell Signaling Technology | 1:1000 | |
| Rabbit p62 antibody | 18420-1-AP | Proteintech | 1:1000 | |
| Rabbit CD44 antibody | 15675-1-AP | Proteintech | 1:200 | 1:1000 |
| Mouse Flag M2 antibody | F1802 | Sigma-Aldrich | 1:1000 | |
| Rabbit Caspase-1 antibody | GB11383 | Servicebio | 1:1000 | |
| Rabbit ASC antibody | GB113966 | Servicebio | 1:1000 | |
| Goat OPN antibody | ab11503 | Abcam | 1:200 | |
| Accession Number | Genes | Primer Sequence (5′–3′) |
|---|---|---|
| NM_00141184 3.1 | GAPDH (112 bp) | F primer: ATACGGCTACAGCAACAGGG R primer: GCCTCTCTTGCTCAGTGTCC |
| NM_023258.4 | ASC (106 bp) | F primer: CTAGTTTGCTGGGGAAAGAAC R primer: CTAAGCACAGTCATTGTGAGCTC |
| NM_009807.2 | Caspase-1 (72 bp) | F primer: CGTACACGTCTTGCCCTCAT R primer: AACTTGAGCTCCAACCCTCG |
| XM_006498- 649.2 | CD44 (191 bp) | F primer: ATGAAGTTGGCCCTGAGCAA R primer: TCTTCTTCAGGAGGGGCTGA |
| NM_007798.3 | CTSB (103 bp) | F primer: GCAGCCAACTCTTGGAACCTT R primer: GGATTCCAGCCACAATTTCTG |
| NM_010705.3 | Galectin-3 (73 bp) | F primer: TTGAAGCTGACCACTTCAAGGTT R primer: AGGTTCTTCATCCGATGGTTGT |
| NM_008361.4 | IL-1β (165 bp) | F primer: CTCACAAGCAGAGCACAAGC R primer: AGCTGTCTGCTCATTCACGA |
| NM_001317- 353.1 | LAMP1 (88 bp) | F primer: TCGTGAACATTTCCCTGCCA R primer: GTGAGGCTGGGGTCAGAAAC |
| XM_00653075 1.4 | MMP-2 (96 bp) | F primer: CCTGGACCCTGAAACCGTG R primer: TCCCCATCATGGATTCGAGAA |
| NM_0013199- 86.1 | MMP-7 (93 bp) | F primer: CTTACCTCGGATCGTAGTGGA R primer: CCCCAACTAACCCTCTTGAAGT |
| NM_004994.3 | MMP-9 (149 bp) | F primer: AAGGGTACAGCCTGTTCCTGGT R primer: CAGGATGCCGTCTATGTCGTC |
| NM_145827.4 | NLRP3 (83 bp) | F primer: ACGAGTCCTGGTGACTTTGTAT R primer: TAGGTCCACACAGAAAGTTCTCTTA |
| NM_001204- 203.1 | OPN (221 bp) | F primer: AGCCACAAGTTTCACAGCCACAAG R primer: CTGAGAAATGAGCAGTTAGTATTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gai, C.; Zhao, Y.; Xin, D.; Li, T.; Cheng, Y.; Jiang, Z.; Song, Y.; Liu, D.; Wang, Z. Mechanistic Insights into the Role of OPN in Mediating Brain Damage via Triggering Lysosomal Damage in Microglia/Macrophage. Cells 2023, 12, 854. https://doi.org/10.3390/cells12060854
Gai C, Zhao Y, Xin D, Li T, Cheng Y, Jiang Z, Song Y, Liu D, Wang Z. Mechanistic Insights into the Role of OPN in Mediating Brain Damage via Triggering Lysosomal Damage in Microglia/Macrophage. Cells. 2023; 12(6):854. https://doi.org/10.3390/cells12060854
Chicago/Turabian StyleGai, Chengcheng, Yijing Zhao, Danqing Xin, Tingting Li, Yahong Cheng, Zige Jiang, Yan Song, Dexiang Liu, and Zhen Wang. 2023. "Mechanistic Insights into the Role of OPN in Mediating Brain Damage via Triggering Lysosomal Damage in Microglia/Macrophage" Cells 12, no. 6: 854. https://doi.org/10.3390/cells12060854