1. Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron (MN) disease [
1]. It is characterized by the progressive degeneration of upper and lower motor neurons (MNs), leading to muscle atrophy, weakness, and spasticity [
2]. Respiratory muscle denervation is typically the cause of death, and it usually occurs within five years of the onset of the disease. Of ALS cases, 90% are sporadic, while 10% are inherited [
2] and are most frequently associated with mutations in SOD1, TARDBP, FUS, and C9ORF72 gene sequences.
The clinical similarities between the sporadic and familial forms of ALS imply that understanding the mechanisms responsible for familial ALS could shed light on both types of the disease [
2].
The notion that MN death leads to ALS is commonly acknowledged. Nonetheless, some ambiguity remains about the sequence in which MN degeneration occurs in ALS. Several investigations in mouse models have demonstrated malfunction or decay of the neuromuscular junction preceding MN loss [
3,
4]. Additionally, distal axonopathy has been observed to occur before neuronal degeneration and disease onset [
5,
6,
7]. Muscle atrophy precedes this sequence of events [
8,
9]. Indeed, the selective expression of mutant SOD1 in the skeletal muscles of mice is sufficient to induce ALS neurodegeneration, suggesting an intrinsic muscle pathology directly impacting MNs [
10,
11].
The inflammatory response differs in its contribution in the peripheral and central nervous system. While the activation of aberrant glial cells, infiltration of T cells, and release of proinflammatory molecules result in neurodegeneration in the CNS, the coordinated efforts of immune cells, including the removal of cellular debris and the release of wound-healing factors, are crucial for successful axon and muscle regeneration [
12,
13]. This may explain the association between higher peripheral inflammation and slower disease progression in mutant SOD1 ALS patients [
14].
We recently showed that macrophage (MΦ) muscle infiltration is pivotal in driving the generation of new myofibers, lessening denervation atrophy and improving motor ability in ALS mouse models [
15].
Muscle regeneration depends on satellite cells (SCs) placed on the muscle fiber surface [
16]. Upon muscle damage, SCs are triggered to exit quiescence and undergo proliferation. Postmitotic myogenic precursor cells resulting from SCs then form multinucleated myotubes, leading to terminal myofiber differentiation and growth. During this process, MΦs are the most abundant immune cell recruited within the damaged tissue where they directly interplay with SCs and orchestrate their fate through different cues [
17,
18,
19]. Among them, IL-10 is a potent immunomodulatory factor able to autoregulate the inflammatory response [
20]. Indeed, IL-10 promotes the M1 to M2 shift of MΦs, which is a mandatory step to gaining steady muscle growth and regeneration [
17]. Furthermore, IL-10 directly affects SC differentiation, escorting the transition of myogenesis from the proliferative to the differentiation stage in the injured muscle [
21].
In this study, we evaluated the possibility of modulating the MΦ fingerprint and sustaining SC conversion to mature myofibers within the skeletal muscle of ALS mouse models. We showed that intramuscular IL-10 injections in transgenic SOD1G93A mice counteract skeletal muscle atrophy, directly stimulating SC differentiation and MΦ polarization. This eventually led to MN preservation and improved the motor performance of transgenic mice.
2. Materials and Methods
2.1. Animals
Transgenic SOD1G93A male and female mice with C57BL/6J (C57-SOD1G93A) and 129S2/Sv (129Sv-SOD1G93A) genetic backgrounds and their corresponding non-transgenic (Ntg) littermates were used in this study [
22,
23]. The animals were housed under SPF (specific-pathogen-free) standard conditions (22 ± 1 °C, 55 ± 10% relative humidity, and a 12 h light/dark schedule). There were 3–4 mice per cage, and the animals had free access to food (standard pellets, Altromin, MT, Rieper) and water.
The tibialis anterior (TA), gastrocnemius medialis (GCM) and quadriceps (QC) were treated daily with recombinant mouse IL-10 (PeproTech, London, UK) at a dose of 0.25 μg/day (10 μL/muscle) as previously described [
24]. Procedures involving animals and their care were conducted in conformity with the institutional guidelines of the Mario Negri Institute for Pharmacological Research (IRFMN), Milan, Italy. The IRFMN adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A, issued 6 March 2008 by the Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorization for persons conducting animal experiments (Quality Management System Certificate– UNI EN ISO 9001:2015, Reg. N° 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE).
2.2. Behavioral Analysis
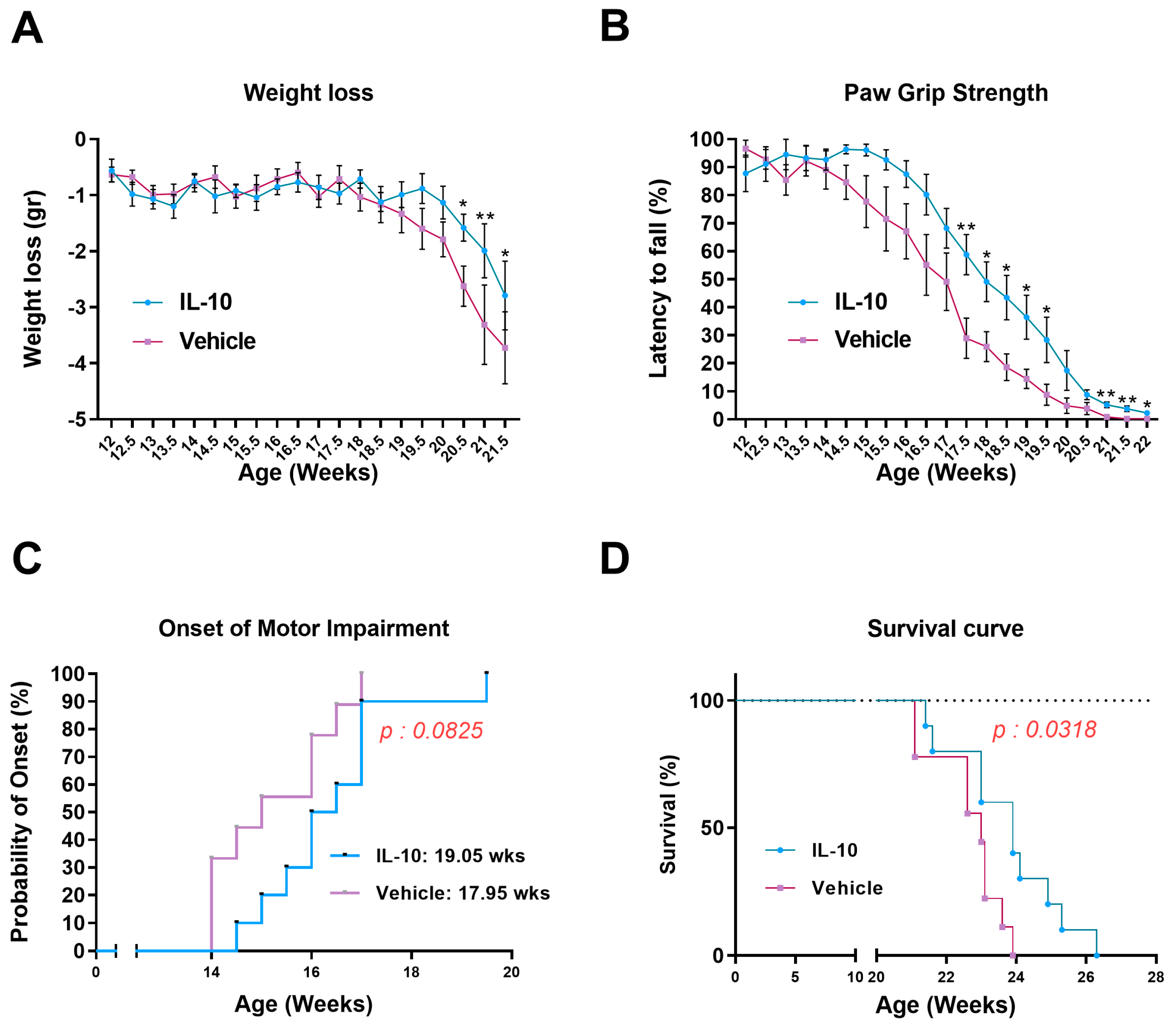
The longitudinal bodyweight loss was determined by dividing the weight of a single mouse at various time points by its maximum weight achieved. For the paw grip strength test, the mouse was placed on the wire lid of a conventional housing cage and gently pulled by the tail until it grasped the grid with all four paws. The lid was then gently turned upside down, and the latency time for the mouse to fall on the table was recorded up to a maximum of 90 s. Each mouse received up to three attempts, and the longest latency was recorded. The onset of motor deficit was considered when the mice showed the first signs of impairment (a latency less than 90 s) in the paw grip strength test. The disease end stage was defined as the point at which the animals could not right themselves within 10 s after being placed on each side. This time point was considered the measure of survival length.
2.3. Dissection of Skeletal Muscles
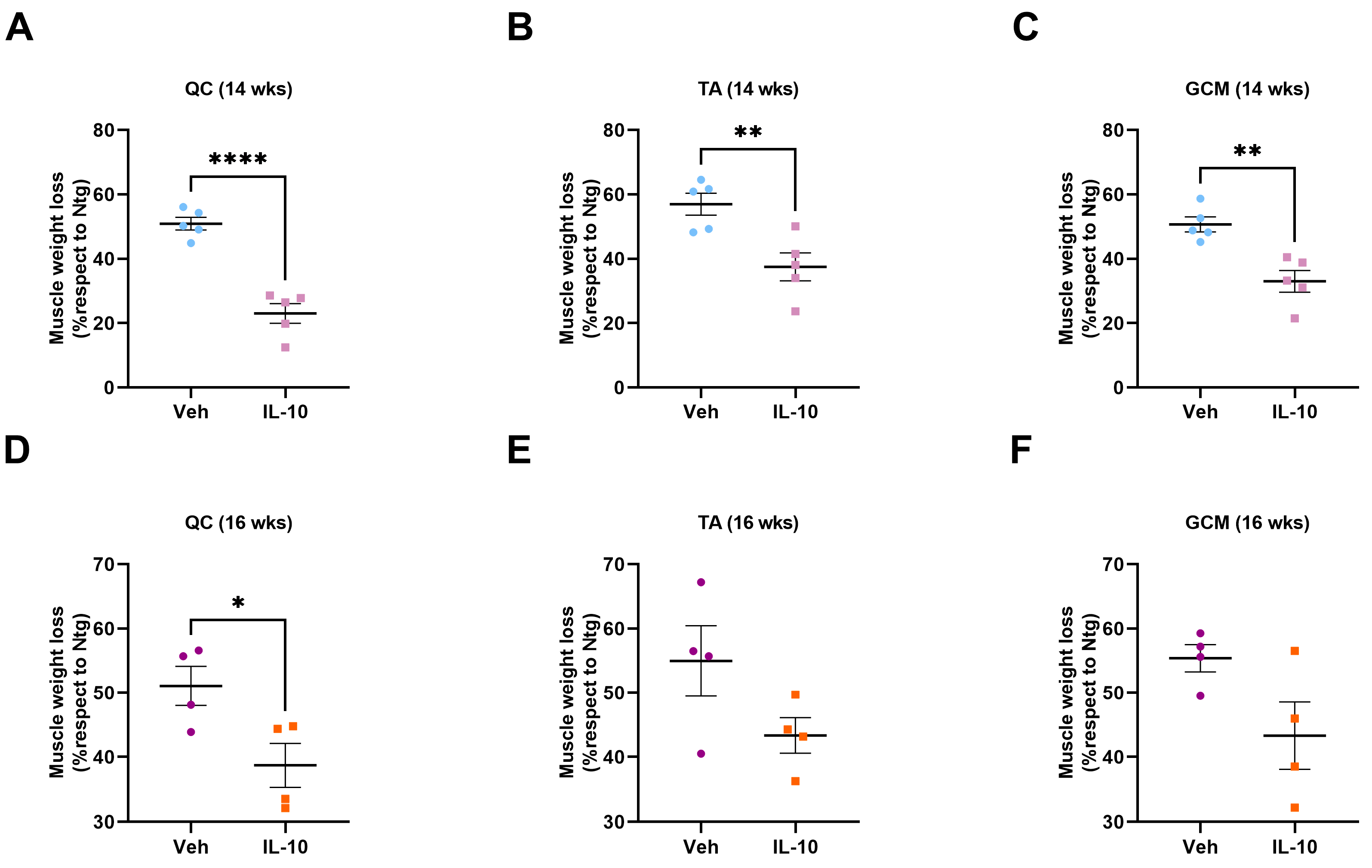
TA, GCM, and QC muscles were collected from C57-SOD1G93A and 129Sv-SOD1G93A male mice and their relative Ntg littermates at 14 and 16 weeks of age, corresponding to the symptom onset (OS) and symptomatic stages based on the performance on the grip strength test. All mice and their relative Ntg littermates were intracardially perfused with 0.1 M PBS. Muscles from each mouse were dissected, snap-frozen in isopentane, cooled in liquid nitrogen, and stored at −80 °C until use. After weighing the TA, GCM, and QC muscles, only the TA muscles were used for immunohistochemical, biochemical, and biomolecular analyses.
2.4. Immunohistochemistry
The tissue dissection process involved freezing the TA with cooled isopentane and leaving the spinal cord in 4% PFA overnight at 4 °C. It was then rinsed and stored for 24 h in 30% sucrose and 0.01 M PBS. For the muscles, 20 μm longitudinal and 10 μm transversal serial TA cryo-sections were collected on poly-lysine objective slides (VWR International, Milan, Italy).
To determine the cross-sectional area (CSA) and fiber type, the sections were incubated with MyHC type I (BA-D5, 1:10; DSHB, Iowa City, IA, USA), MyHC type IIa (SC-71,1:17; DSHB, Iowa City, IA, USA), MyHC type IIb (BF-F3, 1:9; DSHB, Iowa City, IA, USA), Rabbit anti-Laminin (1:100, L9393; Sigma-Aldrich, St. Louis, MO, USA), primary antibodies and respective secondary antibodies, anti-MIgG2b Alexa-flour 564 (A21144,1:500) (Invitrogen, Waltham, MA, USA), anti-MIgG1 Alexa-flour 488 (A21121, 1:500) (Invitrogen, Waltham, MA, USA), anti-MIgM Alexa-flour 647 (A21046,1:500; Invitrogen, Waltham, MA, USA), and anti-Rabbit Alexa Fluor 405 (ab175649, 1:500; Abcam, Cambridge, UK).
To assess the polarization of macrophages, longitudinal muscle TA sections were treated with acetone for 10 min, air-dried, and then washed. The muscle slides were blocked with 10% NGS or NDS in PBS for 1 h and then incubated overnight with primary antibodies: anti-CD11b, rat (1:200; Bio-Rad, Hercules, CA, USA); anti-iNOS, rabbit (1:200; Invitrogen); anti-mannose receptor, rabbit (1:500; Abcam, Cambridge, UK) at 4 °C. Secondary antibodies were as follows: Alexa488, anti-rat and Alexa647, anti-rabbit (1:500; Thermo Fisher, Waltham, MA, USA). The nuclei were counterstained with Hoechst (1:1000; Roche, Basel, Switzerland).
The spinal cord was cut into 30 µm serial transverse sections. The following primary antibodies and staining were used: anti-GFAP, mouse (1:2500; Merck, Darmstadt, Germany); anti-Iba1, rabbit (1:200; Fujifilm Wako Chemicals, Richmond, VA, USA); anti-ChAT, goat (1:200; Sigma-Aldrich, St. Louis, MO, USA). Secondary antibodies used were Alexa488 anti-mouse, Alexa647 anti-rabbit (1:500; Thermo Fisher, Waltham, MA, USA), and Alexa647 anti-goat (1:500; Thermo Fisher, Waltham, MA, USA).
The cells were treated with 4% PFA in PBS for 15 min to fix them, followed by permeabilization with 0.1% Triton for 5 min. To prevent unspecific signals, the cells were blocked with 1% BSA for 30 min. The primary antibodies anti-MF20, mouse (1:50; DSHB, Iowa City, IA, USA); anti-CD11b, rat (1:200; Bio-Rad, Hercules, CA, USA); anti-mannose receptor, rabbit (1:500; Abcam, Cambridge, UK); and anti-Ki67, rabbit (1:200; Abcam, Cambridge, UK) were diluted in blocking solution and incubated overnight at 4 °C. The cells were incubated with the secondary antibodies Alexa488 anti-rabbit, Alexa488 anti-mouse, and Alexa647 anti-rat (1:500; Thermo Fisher, Waltham, MA, USA) for 1 h at room temperature and washed with PBS. Nuclei were counterstained with Hoechst (1:1000; Roche, Basel, Switzerland) in PBS, and glasses were mounted in Fluorsave Mountant (Merck, Darmstadt, Germany). Images were captured using an A1 Nikon confocal microscope at 20× or 40× magnification in sequential scanning mode, and NIS Elements software 4.5 (Nikon, Melville, NY, USA) was used for data acquisition.
All the immunofluorescences were verified with control experiments using only the secondary antibody.
2.5. Real-Time PCR
RNA was extracted from the TA muscles using Trizol (Invitrogen, Waltham, MA, USA) and purified with PureLink RNA columns (Life Technologies, Carlsbad, CA, USA). The RNA samples were then treated with DNase I and reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA). A real-time PCR was carried out on the cDNA specimens in triplicate, using the Taq Man Gene expression assay (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions, with 1x Universal PCR master mix (Life Technologies, Carlsbad, CA, USA) and 1x mix containing specific receptor probes (Life Technologies, Carlsbad, CA, USA). Relative quantification was calculated by determining the ratio between the cycle number (Ct) at which the signal crossed a threshold set within the logarithmic phase of the target gene and that of the reference β-actin gene (4310881E; Life Technologies, Carlsbad, CA, USA). The mean values of the triplicate results for each animal were used as individual data for a 2-ΔCt statistical analysis. The probes used for the real-time PCR were tumor necrosis factor-alpha (TNF-alpha) (Mm00443258_m1; Life Technologies, Carlsbad, CA, USA), interleukin 1β (Il-1β; Mm00434228_m1; Life Technologies, Carlsbad, CA, USA), and interleukin 10 (Il-10; Mm00439614_m1; Life Technologies, Carlsbad, CA, USA).
2.6. Western Blotting
The mice were perfused with 0.1 M PBS, and their tissues were rapidly dissected. The TA tissues were snap-frozen using cooled isopentane and stored at −80 °C until needed. To prepare the samples for analysis, equal amounts of total protein homogenates were loaded onto a polyacrylamide gel and electroblotted onto a PVDF membrane (Millipore, Burlington, MA, USA), following the method previously described in [
25]. The membranes were then immunoblotted with primary antibodies, followed by HRP-conjugated secondary antibodies from Santa Cruz Biotechnology (Dallas, TX, USA). The Luminata Forte Western Chemiluminescent HRP Substrate from Millipore (Burlington, MA, USA) was used to develop the blots, and the Chemi-Doc XRS system from Bio-Rad (Hercules, CA, USA) was used for visualization. The immunoreactivity was normalized to the total amount of protein loaded, as determined by Ponceau staining. The primary antibodies used in the experiment were mouse anti-Pax7 (1:1000; DSHB, Iowa City, IA, USA), rabbit anti-MyoD (1:1000; Proteintech, Rosemont, IL, USA), mouse anti-MyoG (1:130; DSHB, Iowa City, IA, USA), and CD206 (1:500; Abcam, Cambridge, UK).
2.7. Primary Satellite Cell Cultures and Immunofluorescence
The process for isolating and labeling satellite cells was performed following the methods described in Fabbrizio et al. [
25]. Hindlimb muscles were taken from three-week-old mice and digested for 45 min at 37 °C in PBS (Sigma-Aldrich, St. Louis, MO, USA) supplemented with Dispase II (2.4 U/mL, Roche, Basel, Switzerland), Collagenase A (2 mg/mL, Roche, Basel, Switzerland), 0.4 mM CaCl
2, 5 mM MgCl
2, and DNase I (10 μg/mL, Roche, Basel, Switzerland). The resulting cell suspension was filtered and stained with CD45/CD31/Ter119 phycoerythrin (PE) for lineage exclusion, Sca1-FITC (stem cell antigen 1), fluorescein isothiocyanate (FITC), and α7 integrin allophycocyanin (APC) APC. The SCs were then sorted using Moflo Astrios (Beckman Coulter, Brea, CA, USA), seeded onto Matrigel-coated plates (Corning, Corning, NY, USA) at a low density (3500 cells/cm
2), and cultured for four days in Cyto-Grow complete medium (Resnova, Rome, Italy) as a growth medium (GM).
Once the cells reached confluence, they were shifted to a differentiation medium consisting of DMEM supplemented with 2% horse serum and cultured for a further 48 h to induce myogenic differentiation.
SC proliferation was evaluated for each well on image fields acquired with an Olympus virtual slide system VS110 (Olympus, Tokyo, Japan) by counting the number of DAPI+/Ki67+ (Anti-Ki67 Ab; Abcam) cells per field. SC differentiation was assessed by evaluating fiber dimension and the fusion index (%) given by the number of nuclei per myotubes stained with anti-MyHC (DSHB, Iowa City, IA, USA).
For both procedures, each well was acquired applying a stereological random sampling procedure. Briefly, a square grid of sample fields was drawn on the well image using the “Grid” function to ensure that each part of the well was equally likely to be sampled. The analysis was conducted on the average of four image fields acquired at fixed distances from each other.
2.8. Primary Mф Cultures
Primary Mф cultures were obtained as previously described [
25]. The spleen was harvested and mechanically dissociated in RCB buffer (NH
4Cl 150 mM, NaHCO
3 10 mM, and EDTA 1 mM) to obtain a single-cell suspension. The cells were then plated at a concentration of 4 × 10
6 cells/mL in RPMI supplemented with 10% fetal bovine serum, 100 U/mL gentamycin, 100 µg/mL streptomycin, and 100 U/mL penicillin. After 2 h, non-adherent cells were removed, and the medium was enriched with 10 ng/mL mouse macrophage colony-stimulating factor (Sigma-Aldrich, St. Louis, MO, USA). After one week of culture, the cells were used for Western blotting and an immunofluorescence analysis. For MΦ polarization, the cells were stimulated for 24 h with IL-4 (PeproTech, London, UK) (10 ng/mL) or to an M2 phenotype with IL-4 (PeproTech, London, UK) (10 ng/mL) coupled with IL-10 (PeproTech, London, UK) (50 ng/mL) in the presence or absence of an Anti-IL-10 antibody (AbαIL-10; PeproTech, London, UK) (2.5 μg/mL).
2.9. Mф Proliferation Assay
Cell proliferation was analyzed by trypan blue exclusion. Briefly, macrophages were stimulated with IL-10 (PeproTech, London, UK) (50 ng/mL) in the presence or absence of Anti-IL-10 antibody (PeproTech, London, UK) (2.5 μg/mL). After 24 h, the cells were collected and centrifuged for 5 min at 300× g. The resulting pellet was resuspended in 1 mL of PBS. A solution of 0.4% trypan blue was prepared and mixed with 10 µL of the cell suspension for a manual cell count. The counting procedure was repeated at least three times.
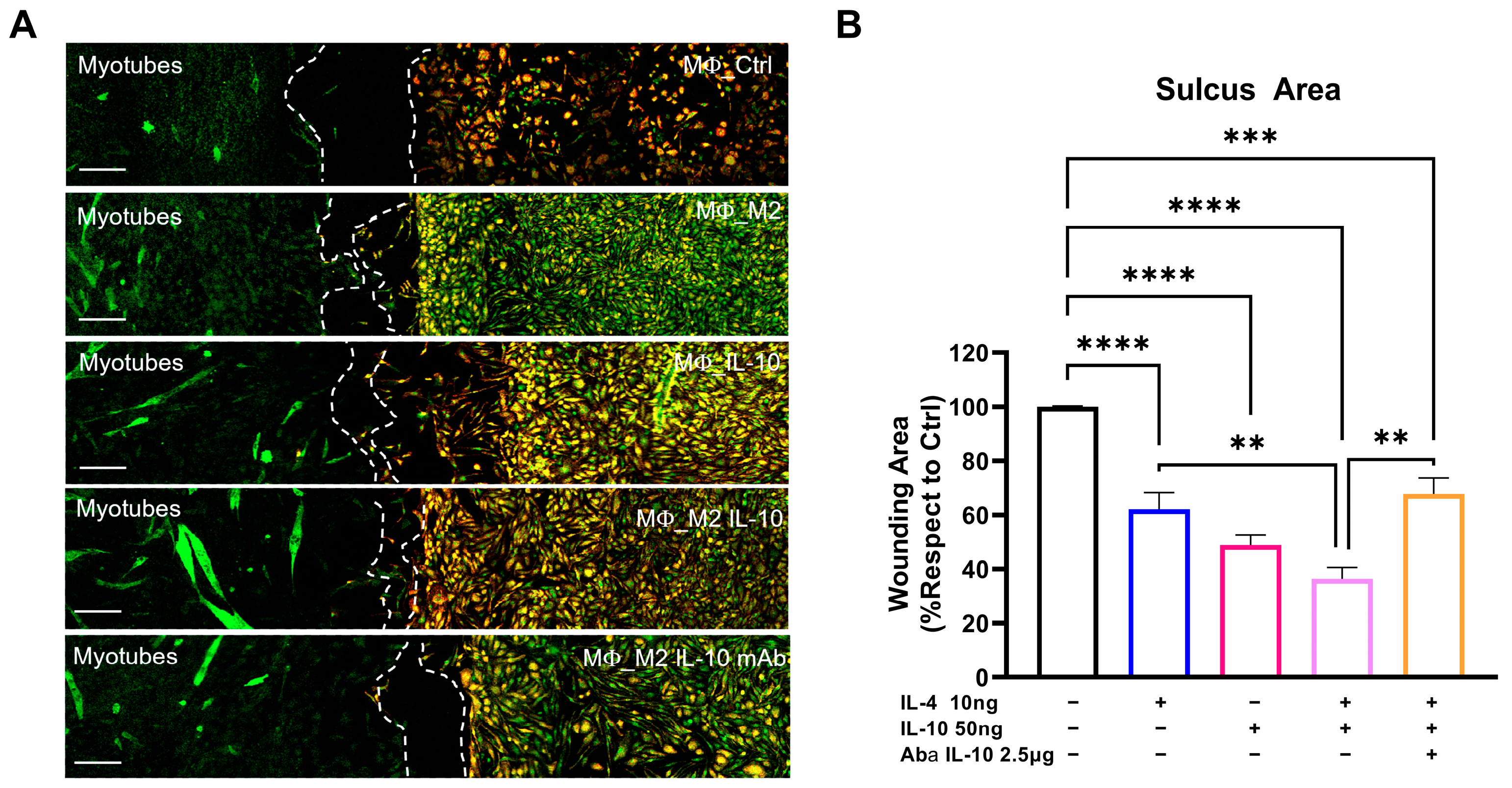
2.10. Sc/Mф Co-Culture for Migration Analysis
For co-culture experiments, the SCs collected from C57-SOD1G93A mice were plated in one sulcus of the Culture-Insert 2 Well (Ibidi, Glasgow, UK), a two-well silicone insert with a defined cell-free gap that is suitable for wound healing, migration assays, 2D invasion assays, and the co-cultivation of cells. SCs were plated at a concentration of 3500 cells/cm2 in GM, which was replaced by DM after 24 h. The following day, the MΦs collected from the spleen of C57-SOD1G93A mice were plated in the other well at a concentration of 15,000 cells/cm2 in DM + M-CSF (1 μg/mL). After 24 h, the MΦs were olarized to an M2 phenotype with IL-4 (10 ng/mL) coupled with an IL-10 (50 ng/mL) treatment in the presence or absence of Anti-IL-10 antibody (2.5 μg/mL). The Culture-Insert that divided the two cultures was removed after 24 h, and the medium was changed with DM + M-CSF to maintain the differentiation of the SCs and to simultaneously support the survival of the MΦs. After 72 h, the cells were fixed with 4% paraformaldehyde and stored in PBS at 4 °C until immunohistochemical analysis.
The migration of the CD11b+ MΦs toward the MF20+ SCs was evaluated using a confocal Nikon A1 running NIS Elements (Nikon, Melville, NY, USA) at 40× magnification and by calculating the area of the sulcus separating the respective cultures with Image J (NIH, Bethesda, MD, USA). The degree of migration was extrapolated by normalizing the sulcus areas with respect to the control.
2.11. Statistics
Statistical analyses were performed using Prism 9.4.1 for Windows (GraphPad Software Inc., Boston, MA, USA). The mean ± SEM values were reported, with the dependent and group variables named on the y- and x-axes of the graph.
A repeated-measures ANOVA, followed by Sidak’s post-analysis, was used to analyse the parameters of disease progression in the SOD1G93A mice (body weight and PaGe test) by checking for normality in the residual and homoscedasticity, using the Geisser–Greenhouse epsilon to evaluate potential violations. The log-rank Mantel–Cox test was used to analyse symptom onset and survival length, with Kaplan–Meier plots generated. Student’s t-test was used for the statistical analysis of two groups, and a one-way ANOVA, followed by Fisher’s multiple comparison test, was used for more than two groups. The D’Agostino–Pearson omnibus normality test and relative QQ plots were used to assess the assumption of normality. A p < 0.05 was considered statistically significant for all analyses.
4. Discussion
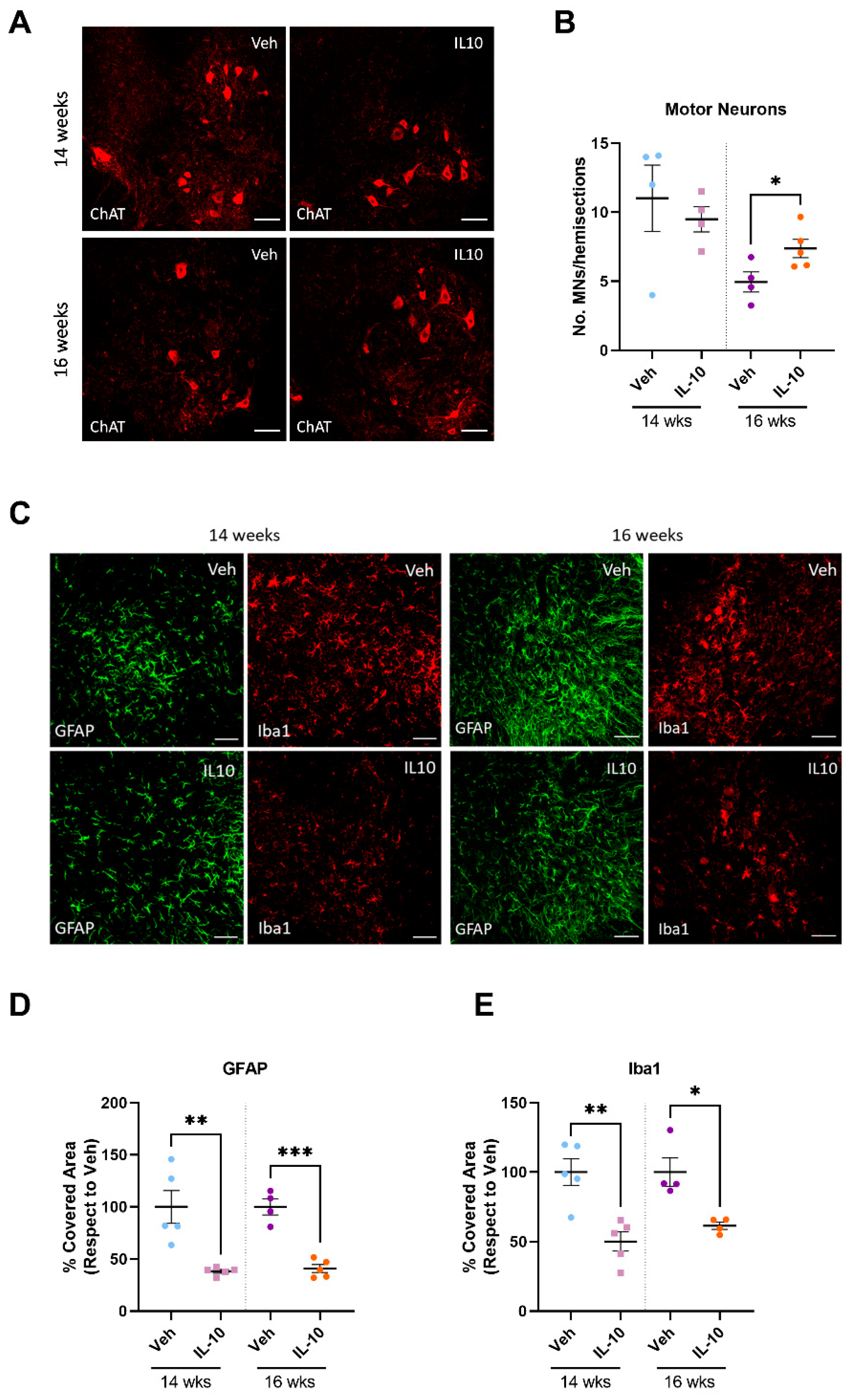
In this study, we established that the intramuscular injection of IL-10 improved the motor function of SOD1G93A mice. The beneficial effect was associated with a straight influence on the SC differentiation and MΦ polarization in the skeletal muscles, and it spread retrogradely to the spinal cord, reducing astrogliosis and preserving MNs.
Efficient SC activation, proliferation, fusion, and differentiation are crucial steps in the myogenic process that enables skeletal muscle to repair itself [
16,
17]. However, research on ALS mice and patients suggests that SCs may be entering a senescent state, disrupting the signaling necessary for optimal myogenesis [
31]. Consequently, skeletal muscles are unable to regenerate, leading to significant muscle atrophy and weakness.
The role of the inflammatory response in muscle repair is critical in terms of both time and space as it serves as a bridge between the initial muscle injury response and muscle reparation [
17,
32]. Immune cells and acute inflammation are essential in almost every stage of muscle regeneration, as evidenced by data from cell biology and immunology. The inflammatory response is dynamic, with Mфs adapting their gene expression programs to control the sequential phases of tissue recovery. Following skeletal muscle injury, proinflammatory Mфs accumulate in the affected tissue area to clear debris and stimulate SC proliferation. Subsequently, these Mфs transition into anti-inflammatory Mфs, also known as pro-regenerative macrophages, which aid in the formation and growth of new myofibers [
17,
32].
We recently demonstrated the pivotal role of the innate, immune-mediated response in preserving skeletal muscle mass and thus the speed of the disease progression in ALS mouse models. The early, self-complementary adeno-associated virus 9 (scAAV9)-mediated monocyte chemoattractant protein 1 (MCP1) boosting in the skeletal muscle of mSOD1 mice significantly enhanced MΦ infiltration, promoting the generation of new myofibers and lessening denervation atrophy and MN loss [
15]. While this approach successfully gave rise to new myofibers in transgenic mice, it did not directly influence the MΦ switch towards a pro-regenerative M2 profile or muscle stem cell differentiation. These processes occurred spontaneously during the disease progression due to the MCP1-mediated increase of MΦ recruitment. Therefore, in this study, we evaluated a preclinical therapeutic approach focused on supporting MΦ transition and triggering SC conversion to mature myofibers by exogenously administering IL-10 in the skeletal muscles of SOD1G93A mice.
IL-10 is a homodimeric type II cytokine composed of 178 amino acids. It binds a heterodimeric receptor (IL-10 receptor, IL-10-R), inducing an anti-inflammatory response [
33]. The cytokine is mainly produced by antigen-presenting cells (APCs), including monocytes/MΦ, which are the main targets of IL-10 immunosuppressive effect as they express higher levels of IL-10R [
20,
34].
In the CSF of ALS patients with mild symptoms and a slow progressive course, IL-10 levels are elevated, suggesting a possible neuroprotective and anti-inflammatory action of this cytokine [
35]. Additionally, preclinical ALS studies reported delays in symptom onset and improved survival in a SOD1G93A mouse model with lifelong AAV-IL-10 overexpression in the spinal cord [
36,
37]. These studies are encouraging in defining a possible therapeutic strategy for ALS. Nevertheless, the effect of IL-10 at the tissue level, especially in peripheral districts, has never been investigated.
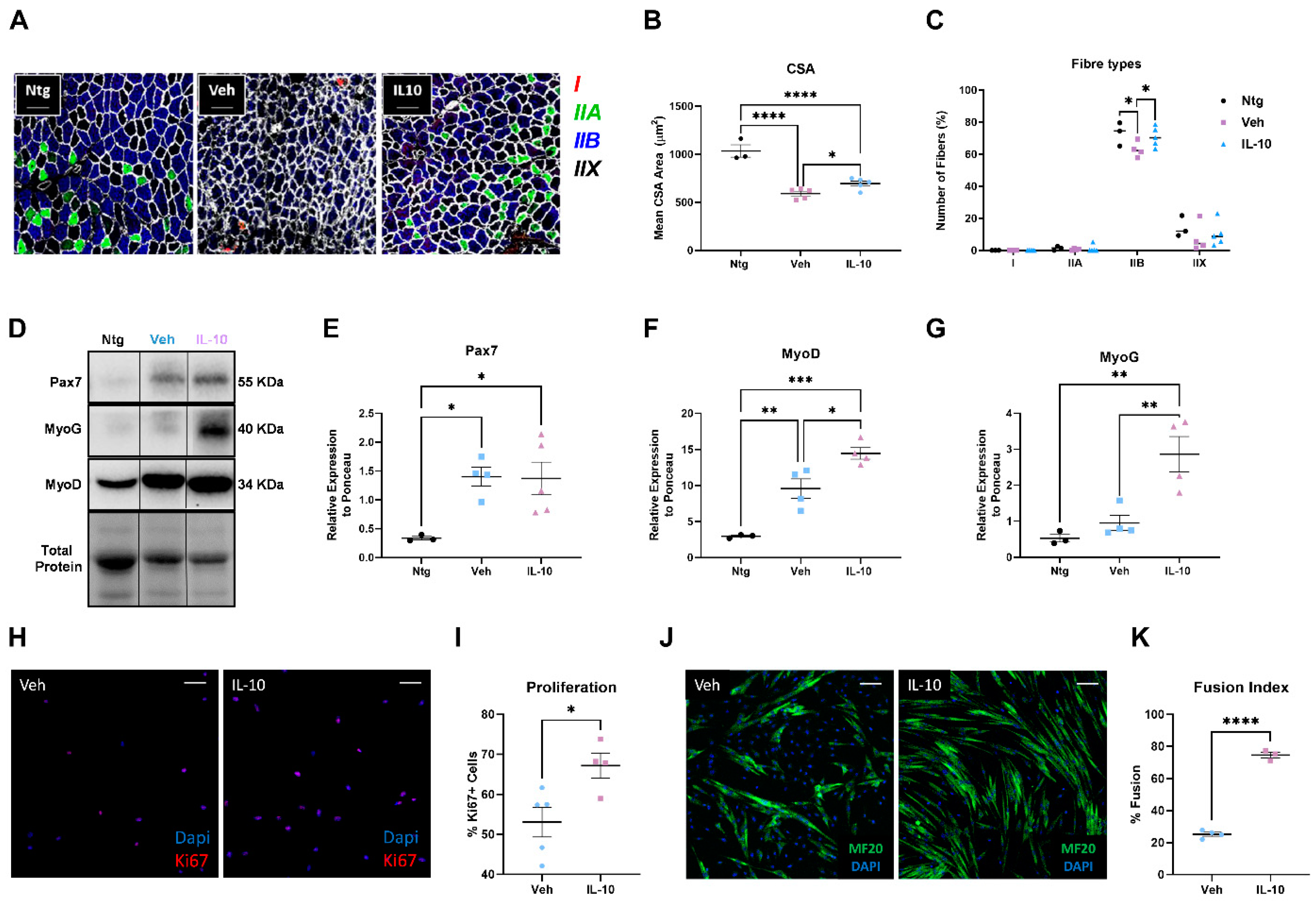
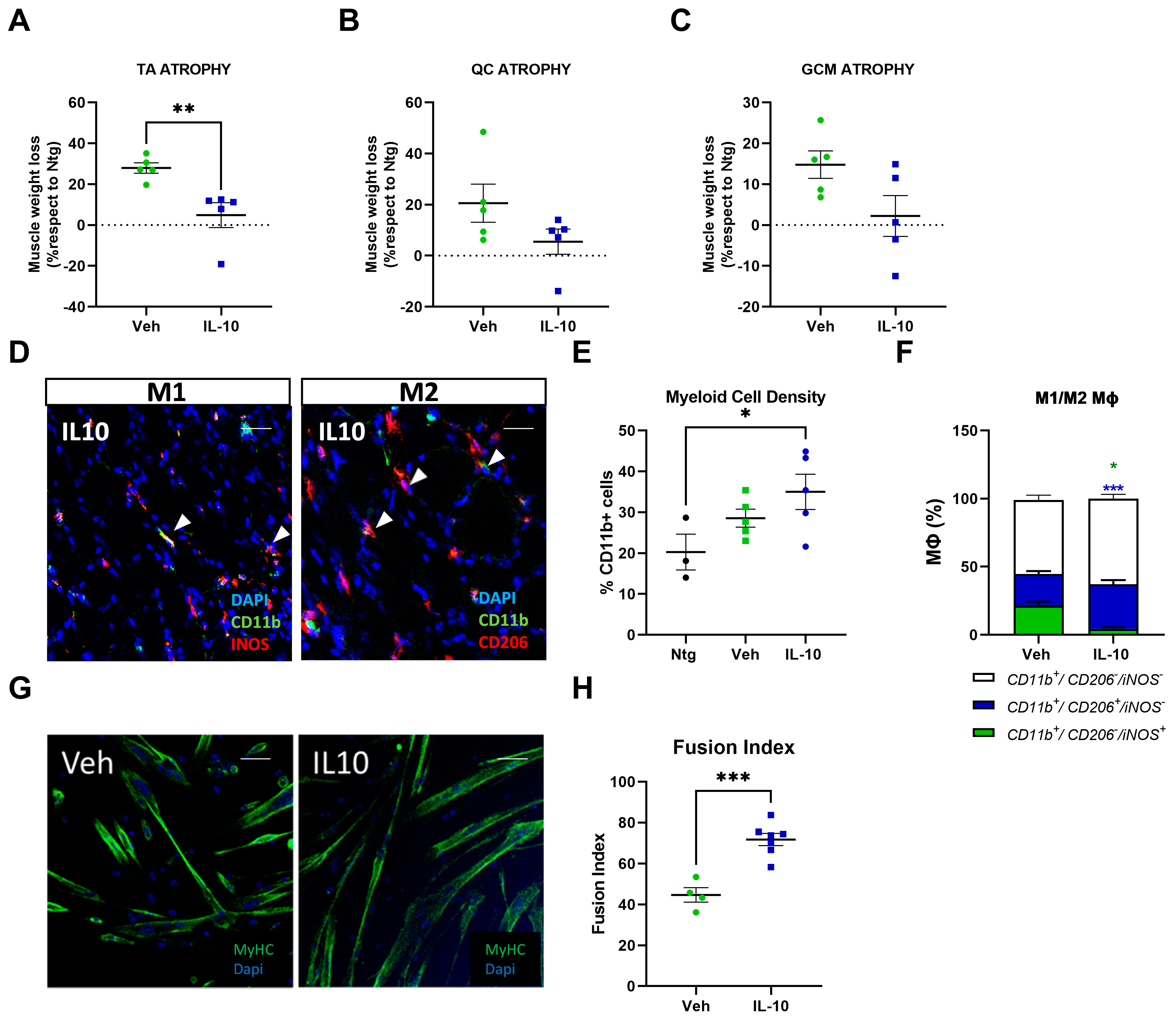
This study demonstrated that the administration of IL-10 delayed hindlimb skeletal muscle atrophy and increased muscle fiber size by enhancing the expression of pro-differentiating factors, MyoD and MyoG. Additionally, SCs derived from the IL-10-treated muscles and cultured ex vivo displayed an improved ability to generate new mature myotubes, indicating the presence of an imprinted memory resulting from environmental cues.
4.1. The IL-10 Signaling Elicited the In Situ Proliferation and Polarization of MΦ toward an M2-Biased Phenotype in the Skeletal Muscle of C57-SOD1G93A Mice
IL-10’s pivotal role in myogenesis is due to its immunomodulatory effects towards APCs. In injured skeletal muscles, IL-10 is expressed by resident and infiltrating MΦs to autoregulate the inflammatory response and escort the transition of myogenesis from the proliferative stage to the differentiation/growth stage [
17,
27,
38]. Specifically, IL-10 inhibits the infiltration of neutrophils and MΦ-M1s into the damaged tissue [
39], limiting the expression of proinflammatory cytokines, and elicits the M1 to M2 shift of MΦs, a mandatory step for gaining regular muscle growth and regeneration.
In a model of chronic skeletal muscle infections, intramuscular injections of IL-10 improved muscle function and skewed MΦs toward a restorative phenotype. These shifts in the MΦ phenotype were coupled with enhanced physiologic parameters of regeneration [
26]. Also, IL-10 influenced the MΦ profile during chronic muscle atrophy. Indeed, treating mdx-derived muscle MΦs with IL-10 lessened the activation of the M1-MΦs in favour of M2-MΦs [
21]. Notably, IL-10 signaling was downregulated in mdx mouse skeletal muscles depleted of MΦs, while ex vivo primary SCs showed differentiation defects in myotube formation. Noteworthy, this imprinting is reversible and can be rescued in vitro by the interplay with MΦ or IL-10 challenging, surmising that MΦs recover SC differentiation defects via IL-10 signaling [
24].
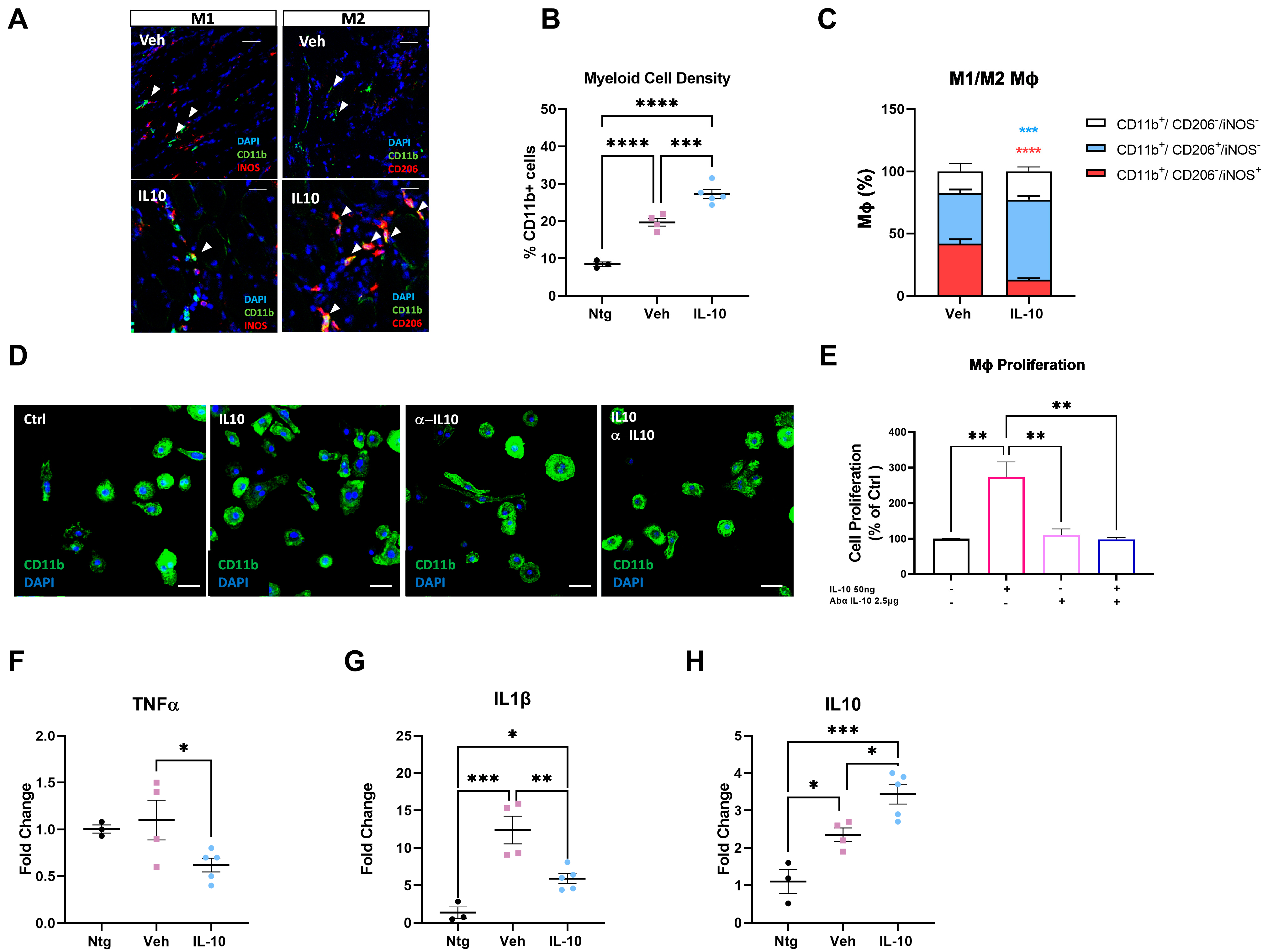
In keeping with this evidence, we found that IL-10 signaling influenced the peripheral immune response within the skeletal muscles of C57-SOD1G93A mice during the generation of new myofibers. It is noteworthy that the exogenous administration of IL-10 increased CD11b
+ cell muscle density, suggesting a direct effect of the cytokine on Mфs in situ proliferation. Indeed, IL-10 administration to primary C57-SOD1G93A Mфs elicited their proliferation index in vitro. This process was previously reported by Jenkins et al. [
40], who revealed that the expansion of innate cells required for pathogen control or wound repair can occur without the recruitment of tissue-destructive inflammatory cells.
Our study found that the intramuscular administration of IL-10 led to an increase in M2-MΦs within the skeletal muscles while reducing the presence of M1-Mфs and related proinflammatory factors, such as IL1β and TNFα. We further investigated the influence of IL-10 on Mфs and found that the co-administration of IL-4 and IL-10 to C57-SOD1G93A-derived Mф cultures increased the M2 profile, as evidenced by the increased expression of CD206. However, IL-10 alone was insufficient in inducing an M2 phenotype in MΦs. Conversely, in vitro polarization shift of Mфs toward the M2 phenotype was significantly suppressed following IL-10 depletion, framing the cytokine as an immunomodulator supporting but not directly eliciting the transition of MΦs to the anti-inflammatory M2 fingerprint.
Muscle IL-10 activity is not exclusive to MΦs, but the IL-10 signaling is pivotal in promoting MΦ-SCs cross-talk, an essential process in immune-cell-mediated myogenesis [
17]. We found that IL-10 is crucial in mediating MΦ displacement in vitro towards SCs regardless of the degree of polarization. Although the mechanism underlying this interaction is still to be elucidated, it is likely that IL-10-treated MΦs elicited the expression of chemo-attractive factors in SCs. Indeed, following muscle insult, SCs upregulated MCP1 to initiate monocyte recruitment and interplay with MΦs to amplify chemotaxis and enhance muscle growth [
28].
4.2. The IL-10 Signaling Triggers Satellite Cell Differentiation in an Immune-Independent Manner
In addition to influencing the inflammatory state of MΦs in the muscle, IL-10 acts directly on the fate of SCs. Indeed, IL-10 administration in primary C57-SOD1G93A SC cultures remarkably boosted their differentiation, which was fully inhibited upon IL-10 depletion. Our results corroborate previous findings that reported IL-10 ablation significantly slowed myogenesis in regenerating muscle during acute injury or chronic disease (i.e., Duchenne Muscular Dystrophy, DMD), showing a lower growth of new muscle fibers and poor resilience to damage [
21,
27,
41]. As a counter-proof, IL-10 injections in MΦ-depleted mdx mice improved the differentiative potential of ex vivo SCs [
24].
We validated this evidence in an ALS-like disease using 129Sv-SOD1G93A mice whose genetic background is associated with defective immune cell recruitment during inflammation [
12,
15,
30]. Intramuscular IL-10 injections in these mice delayed hindlimb muscle atrophy, promoting the pro-regenerative activity of resident MΦs and enhancing the differentiation rate of ex vivo isolated SCs.
4.3. Preservation of Skeletal Muscle Influences Inflammation and Motor Neuron Survival within the Spinal Cord of C57-SOD1G93A Mice
Skeletal muscle plays a significant role in promoting neuron survival, axonal growth, and the maintenance of synaptic connections through anabolic signals and electric impulses. [
42]. After a muscle injury, the altered activity of satellite cells (SCs) releases various cytokines/factors, which are transmitted throughout the body. [
43,
44,
45]. These signals from newly formed myofibers and SCs might be crucial in preventing motor neuron (MN) loss. To investigate if muscle preservation impacts the protection of the entire motor unit, the study extended its analysis beyond skeletal muscles. The results showed a reduction in MN loss in the spinal cord and a decrease in astrocytosis and microgliosis throughout the progression of the disease. It is noteworthy that our approach turned out to be more effective than previous protocols in which the AAV-mediated overexpression of IL-10 in the CNS exacerbated microgliosis [
36] without affecting the extent of MN loss [
37].
5. Conclusions
In conclusion, we demonstrated that the intramuscular injection of IL-10 in SOD1G93A mice improved motor performance by decreasing muscular atrophy and mitigating CNS inflammation and MN loss.
In summary, we found that cytokine activity in the skeletal muscles of transgenic ALS mice had multiple targets. IL-10 signaling was responsible for preserving muscle fiber architecture and delaying muscle atrophy by promoting the differentiation of mature myofibers. This process was directly influenced by the effect of IL-10 on SCs and indirectly impacted by the polarization of MΦs toward an anti-inflammatory and pro-regenerative phenotype. Additionally, our findings showed that improving the condition of the skeletal muscle had a significant impact on the pathology in the CNS. This suggests that MN loss in SOD1G93A transgenic mice may be a form of retrograde, dying-back degeneration following muscle disease and is similar to human ALS [
6].
Despite numerous attempts at developing therapies, the high rate of therapeutic failures has reinforced the notion that ALS is a complex disease involving various factors and systems. The disease is characterized by alterations in the structural, physiological, and metabolic parameters of MNs, glia, and muscles, which act synergistically to worsen the condition. Therefore, therapeutic strategies must target multiple mechanisms and various cells and tissues to be effective.
The encouraging results from this study have broadened our understanding of immune dynamics in the skeletal muscle of ALS mice, providing essential prospects for developing targeted immunomodulatory treatments to be used in conjunction with CNS-targeted drugs to enhance the effectiveness of potential clinical treatments. An exciting perspective could concern the long-term modulation of the peripheral immune system in the skeletal muscle with the use of AAV-mediated therapies.
Our findings afford a possible explanation for the failure of untargeted immunomodulatory treatments in ALS [
46,
47] and suggest that the characterization of the immune muscle profile in patients might be a clinical adjunct to improve the clinical practice and develop innovative and personalized strategies to hinder the disease progression.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






