
Cracking the Code of Neuronal Cell Fate
 ,
,  and
and 
Abstract
:1. Introduction
2. Systems Biology Approaches to Explore the Transcriptional Programs Underlying Neuronal Apoptosis and Survival
3. Apoptosis/Survival Switch and Human Diseases: At the Crossroads between Cancer and Neurodegenerative Diseases
4. Cracking the Transcriptional Regulatory Programs of Neuronal Cell Fate May Orient New Therapeutic Strategies
4.1. Homeobox D9 (Hoxd9)
4.2. Nuclear Receptor 4A1 (Nr4a1)
4.3. Musculoaponeurotic Fibrosarcoma (Maf)
4.4. CCAAT Enhancer Binding Protein Beta (Cebpb)
4.5. Oligodendrocyte Transcription Factor2 (Olig2)
4.6. One Cut Homeobox 2 (Onecut2)
4.7. SAM Pointed Domain Containing ETS Transcription Factor (Spdef)
4.8. Nuclear Transcription Factor Y Subunit Beta (Nfyb)
4.9. Twist Family BHLH Transcription Factor 2 (Twist2)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Erekat, N.S. Apoptosis and its therapeutic implications in neurodegenerative diseases. Clin. Anat. 2022, 35, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Neuronal Life-and-Death Signaling, Apoptosis, and Neurodegenerative Disorders. Antioxid. Redox Signal. 2006, 8, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamada, M.; Nishio, C.; Konishi, A.; Hatanaka, H. SNRK, a member of the SNF1 family, is related to low K(+)-induced apoptosis of cultured rat cerebellar granule neurons. Brain Res. 2000, 873, 274–282. [Google Scholar] [CrossRef]
- Galli, C.; Meucci, O.; Scorziello, A.; Werge, T.M.; Calissano, P.; Schettini, G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: The involvement of intracellular calcium and RNA synthesis. J. Neurosci. 1995, 15, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Valenzulela, E.; Gorczyca, W.; Darzynkiewicz, Z.; Sharma, S.C. Apoptosis in adult retinal ganglion cells after axotomy. J. Neurobiol. 1994, 25, 431–438. [Google Scholar] [CrossRef]
- Martin, D.P.; Schmidt, R.E.; DiStefano, P.S.; Lowry, O.H.; Carter, J.G.; Johnson, E.M. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 1988, 106, 829–844. [Google Scholar] [CrossRef]
- D’Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, S. Cracking the code of neuronal apoptosis and survival. Cell Death Dis. 2015, 6, e1963. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, S. Neuronal apoptosis revealed by genomic analysis: Integrating gene expression profiles with functional information. Neuroinformatics 2007, 5, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, S.; Calissano, P. A Genomic Approach to Investigate Neuronal Apoptosis. Curr. Alzheimer Res. 2006, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, S.; D’Agata, V.; Alessi, E.; Coffa, S.; Alkon, D.L.; Manickam, P.; Ciotti, M.T.; Possenti, R.; Bonini, P.; Marlier, L.; et al. Gene expression profiles of apoptotic neurons. Genomics 2004, 84, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Paratore, S.; Parenti, R.; Torrisi, A.; Copani, A.; Cicirata, F.; Cavallaro, S. Genomic profiling of cortical neurons following exposure to β-amyloid. Genomics 2006, 88, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetmańczyk-Sawicka, K.; Iwanicka-Nowicka, R.; Fogtman, A.; Cieśla, J.; Włodarski, P.; Żyżyńska-Granica, B.; Filocamo, M.; Dardis, A.; Peruzzo, P.; Bednarska-Makaruk, M.; et al. Changes in global gene expression indicate disordered autophagy, apoptosis and inflammatory processes and downregulation of cytoskeletal signalling and neuronal development in patients with Niemann–Pick C disease. Neurogenetics 2020, 21, 105–119. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of Autophagy, Mitophagy, and Apoptosis Genes in the CA3 Region of the Hippocampus in the Ischemic Model of Alzheimer’s Disease in the Rat. J. Alzheimer’s Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Calissano, P.; Matrone, C.; Amadoro, G. Apoptosis and in vitro Alzheimer’s disease neuronal models. Commun. Integr. Biol. 2009, 2, 163–169. [Google Scholar] [CrossRef]
- Parenti, R.; Paratore, S.; Torrisi, A.; Cavallaro, S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during β-amyloid-induced apoptosis. Eur. J. Neurosci. 2007, 26, 2444–2457. [Google Scholar] [CrossRef]
- Estus, S.; Tucker, H.M.; Van Rooyen, C.; Wright, S.; Brigham, E.F.; Wogulis, M.; Rydel, R.E. Aggregated Amyloid-β Protein Induces Cortical Neuronal Apoptosis and Concomitant “Apoptotic” Pattern of Gene Induction. J. Neurosci. 1997, 17, 7736. [Google Scholar] [CrossRef] [Green Version]
- Lossi, L.; Gambino, G. Apoptosis of the cerebellar neurons. Histol. Histopathol. 2008, 23, 367–380. [Google Scholar] [CrossRef]
- Jung, C.G.; Uhm, K.O.; Miura, Y.; Hosono, T.; Horike, H.; Khanna, K.K.; Kim, M.J.; Michikawa, M. Beta-amyloid increases the expression level of ATBF1 responsible for death in cultured cortical neurons. Mol. Neurodegener. 2011, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maino, B.; Ciotti, M.T.; Calissano, P.; Cavallaro, S. Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int. J. Mol. Sci. 2014, 15, 5596–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maino, B.; D’Agata, V.; Severini, C.; Ciotti, M.T.; Calissano, P.; Copani, A.; Chang, Y.C.; Delisi, C.; Cavallaro, S. Igf1 and pacap rescue cerebellar granule neurons from apoptosis via a common transcriptional program. Cell Death Discov. 2015, 1, 15029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.-M.; Paul, S.M. Cultured Cerebellar Granule Neurons as a Model of Neuronal Apoptosis. Apoptosis Tech. Protoc. 1997, 47–66. [Google Scholar] [CrossRef]
- Chiang, L.W.; Grenier, J.M.; Ettwiller, L.; Jenkins, L.P.; Ficenec, D.; Martin, J.; Jin, F.; DiStefano, P.S.; Wood, A. An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proc. Natl. Acad. Sci. USA 2001, 98, 2814–2819. [Google Scholar] [CrossRef] [Green Version]
- Austdal, L.P.E.; Mathisen, G.H.; Løberg, E.M.; Paulsen, R.E. Calcium-induced apoptosis of developing cerebellar granule neurons depends causally on NGFI-B. Int. J. Dev. Neurosci. 2016, 55, 82–90. [Google Scholar] [CrossRef]
- Fernández-Suárez, D.; Krapacher, F.A.; Andersson, A.; Ibáñez, C.F.; Kisiswa, L. MAG induces apoptosis in cerebellar granule neurons through p75NTR demarcating granule layer/white matter boundary. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Weller, M.; Fontana, A. TGF-β-Induced Apoptosis of Cerebellar Granule Neurons Is Prevented by Depolarization. J. Neurosci. 1996, 16, 4174. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, J.; Park, D.S.; Greene, L.A.; Shelanski, M.L. Role of Cell Cycle Regulatory Proteins in Cerebellar Granule Neuron Apoptosis. J. Neurosci. 1999, 19, 8747–8756. [Google Scholar] [CrossRef] [Green Version]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef]
- Williams, R.W.; Herrup, K. The Control of Neuron Number. Annu. Rev. 2003, 11, 423–453. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Di Luzio, A.; Ciotti, M.T.; Calissano, P.; Galli, C. Granule neuron DNA damage following deafferentation in adult rats cerebellar cortex: A lesion model. Neuroscience 1999, 95, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Maino, B.; Spampinato, A.G.; Severini, C.; Petrella, C.; Ciotti, M.T.; D’Agata, V.; Calissano, P.; Cavallaro, S. The trophic effect of nerve growth factor in primary cultures of rat hippocampal neurons is associated to an anti-inflammatory and immunosuppressive transcriptional program. J. Cell. Physiol. 2018, 233, 7178–7187. [Google Scholar] [CrossRef]
- Pattarawarapan, M.; Burgess, K. Molecular basis of neurotrophin-receptor interactions. J. Med. Chem. 2003, 46, 5277–5291. [Google Scholar] [CrossRef]
- Cavallaro, S.; Copani, A.; D’Agata, V.; Musco, S.; Petralia, S.; Ventra, C.; Stivala, F.; Travali, S.; Canonico, P.L. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol. Pharmacol. 1996, 50, 60–66. [Google Scholar] [PubMed]
- Amadoro, G.; Pieri, M.; Ciotti, M.T.; Carunchio, I.; Canu, N.; Calissano, P.; Zona, C.; Severini, C. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: Evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology 2007, 52, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Pamantung, T.F.; Fontaine, M.; Fournier, A.; Vaudry, H. The neuroprotective effect of pituitary adenylate cyclase-activating polypeptide on cerebellar granule cells is mediated through inhibition of the CED3-related cysteine protease caspase-3/CPP32. Proc. Natl. Acad. Sci. USA 2000, 97, 13390–13395. [Google Scholar] [CrossRef] [Green Version]
- Linseman, D.A.; Phelps, R.A.; Bouchard, R.J.; Le, S.S.; Laessig, T.A.; McClure, M.L.; Heidenreich, K.A. Insulin-Like Growth Factor-I Blocks Bcl-2 Interacting Mediator of Cell Death (Bim) Induction and Intrinsic Death Signaling in Cerebellar Granule Neurons. J. Neurosci. 2002, 22, 9287–9297. [Google Scholar] [CrossRef] [Green Version]
- Gleichmann, M.; Weller, M.; Schulz, J.B. Insulin-like growth factor-1-mediated protection from neuronal apoptosis is linked to phosphorylation of the pro-apoptotic protein BAD but not to inhibition of cytochrome c translocation in rat cerebellar neurons. Neurosci. Lett. 2000, 282, 69–72. [Google Scholar] [CrossRef]
- Paratore, S.; Teresa Ciotti, M.; Basille, M.; Vaudry, D.; Gentile, A.; Parenti, R.; Calissano, P.; Cavallaro, S. Gastric inhibitory polypeptide and its receptor are expressed in the central nervous system and support neuronal survival. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 210–222. [Google Scholar] [CrossRef]
- Seaborn, T.; Masmoudi-Kouli, O.; Fournier, A.; Vaudry, H.; Vaudry, D. Protective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) against apoptosis. Curr. Pharm. Des. 2011, 17, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Weller, M.; Klockgether, T. Potassium Deprivation-Induced Apoptosis of Cerebellar Granule Neurons: A Sequential Requirement for New mRNA and Protein Synthesis, ICE-Like Protease Activity, and Reactive Oxygen Species. J. Neurosci. 1996, 16, 4696–4706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, G.; Villari, A.; Spampinato, A.G.; La Cognata, V.; Guarnaccia, M.; Gentile, G.; Ciotti, M.T.; Calissano, P.; D’agata, V.; Severini, C.; et al. Transcriptional profiles of cell fate transitions reveal early drivers of neuronal apoptosis and survival. Cells 2021, 10, 3238. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, L.; Zhang, Z.; Qin, C.; Yang, B.; Ke, Y. TYRP1 Protects Against the Apoptosis and Oxidative Stress of Retinal Ganglion Cells by Binding to PMEL. Ocul. Immunol. Inflamm. 2022. [Google Scholar] [CrossRef]
- Mercurio, D.; Oggioni, M.; Fumagalli, S.; Lynch, N.J.; Roscher, S.; Minuta, D.; Perego, C.; Ippati, S.; Wallis, R.; Schwaeble, W.J.; et al. Targeted deletions of complement lectin pathway genes improve outcome in traumatic brain injury, with MASP-2 playing a major role. Acta Neuropathol. Commun. 2020, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Gangwani, K.; Snigdha, K.; Kango-Singh, M. Tep1 Regulates Yki Activity in Neural Stem Cells in Drosophila Glioma Model. Front. Cell Dev. Biol. 2020, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Srikant, C.B. Cell Cycle Dependent Induction of Apoptosis by Somatostatin Analog SMS 201-995 in AtT-20 Mouse Pituitary Cells. Biochem. Biophys. Res. Commun. 1995, 209, 400–406. [Google Scholar] [CrossRef]
- Stumm, R.K.; Zhou, C.; Schulz, S.; Endres, M.; Kronenberg, G.; Allen, J.P.; Tulipano, G.; Höllt, V. Somatostatin Receptor 2 Is Activated in Cortical Neurons and Contributes to Neurodegeneration after Focal Ischemia. J. Neurosci. 2004, 24, 11404. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Huang, C.; Liu, J.; Shi, X.; Li, X. The significance of PAK4 in signaling and clinicopathology: A review. Open Life Sci. 2022, 17, 586–598. [Google Scholar] [CrossRef]
- Civiero, L.; Greggio, E. PAKs in the brain: Function and dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 444–453. [Google Scholar] [CrossRef]
- Gnesutta, N.; Qu, J.; Minden, A. The Serine/Threonine Kinase PAK4 Prevents Caspase Activation and Protects Cells from Apoptosis. J. Biol. Chem. 2001, 276, 14414–14419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, C.; Liang, W.; Zhang, C.; Wang, Y.; Yang, Y.; Wang, X.; Wang, S.; Huo, D.; Wang, H.; Wang, D.; et al. PAK4 suppresses motor neuron degeneration in hSOD1G93A-linked amyotrophic lateral sclerosis cell and rat models. Cell Prolif. 2021, 54. [Google Scholar] [CrossRef] [PubMed]
- Pütz, S.M.; Kram, J.; Rauh, E.; Kaiser, S.; Toews, R.; Lueningschroer-Wang, Y.; Rieger, D.; Raabe, T. Loss of p21-activated kinase Mbt/PAK4 causes Parkinson-like phenotypes in Drosophila. Dis. Model. Mech. 2021, 14, dmm047811. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Park, M.H.; You, S.T.; Choi, S.W.; Kim, H.K.; McLean, C.; Bae, S.C.; Kim, S.R.; Jin, B.K.; Lee, K.H.; et al. Nigral dopaminergic PAK4 prevents neurodegeneration in rat models of Parkinson’s disease. Sci. Transl. Med. 2016, 8, 367ra170. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Park, J.J.; Shin, E.Y.; Kim, E.G. PAK4 signaling in health and disease: Defining the PAK4–CREB axis. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arévalo, J.C.; Waite, J.; Rajagopal, R.; Beyna, M.; Chen, Z.Y.; Lee, F.S.; Chao, M.V. Cell Survival through Trk Neurotrophin Receptors Is Differentially Regulated by Ubiquitination. Neuron 2006, 50, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Majdan, M.; Walsh, G.S.; Aloyz, R.; Miller, F.D. TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J. Cell Biol. 2001, 155, 1275. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Gerling, N.; Lehmann, M.; Nikolova-Karakashian, M.; Prehn, J.H.M.; Mattson, M.P.; Krieglstein, J. Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor p75. Neuroscience 2002, 115, 1089–1108. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, T.J.; Xiong, Z.Q. NGF-dependent retrograde signaling: Survival versus death. Cell Res. 2009, 19, 525–526. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Moheban, D.B.; Conway, B.R.; Bhattacharyya, A.; Segal, R.A. Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J. Neurosci. 2000, 20, 5671–5678. [Google Scholar] [CrossRef] [Green Version]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Behl, C. Mechanisms of neurodegenerative disorders: Part 2: Control of cell death. Arch. Neurol. 2000, 57, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhao, S.; Li, Y.; Zhang, D.; Wang, B.; Xie, J.; Wang, J. Regulated cell death: Discovery, features and implications for neurodegenerative diseases. Cell Commun. Signal. 2021, 19, 1–29. [Google Scholar] [CrossRef]
- Tatton, W.G.; Olanow, C.W. Apoptosis in neurodegenerative diseases: The role of mitochondria. Biochim. Biophys. Acta 1999, 1410, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe, E.; Wix, R. Neuronal migration, apoptosis and bipolar disorder. Rev. Psiquiatr. Salud Ment. 2012, 5, 127–133. [Google Scholar] [CrossRef]
- Karlović, D. Apoptosis—The potential pathophysiological mechanism in mood disorders modifiable by lithium salts. Biochem. Med. 2008, 18, 291–310. [Google Scholar] [CrossRef]
- Margolis, R.L.; Chuang, D.M.; Post, R.M. Programmed cell death: Implications for neuropsychiatric disorders. Biol. Psychiatry 1994, 35, 946–956. [Google Scholar] [CrossRef]
- Andreone, B.J.; Larhammar, M.; Lewcock, J.W. Cell Death and Neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a036434. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, S.R.; Chin, P.C. Treating neurodegenerative conditions through the understanding of neuronal apoptosis. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Parul; Mishra, A.; Singh, S.; Singh, S.; Tiwari, V.; Chaturvedi, S.; Wahajuddin, M.; Palit, G.; Shukla, S. Chronic unpredictable stress negatively regulates hippocampal neurogenesis and promote anxious depression-like behavior via upregulating apoptosis and inflammatory signals in adult rats. Brain Res. Bull. 2021, 172, 164–179. [Google Scholar] [CrossRef]
- Bachis, A.; Cruz, M.I.; Nosheny, R.L.; Mocchetti, I. Chronic Unpredictable Stress Promotes Neuronal Apoptosis in the Cerebral Cortex. Neurosci. Lett. 2008, 442, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, A.; Gan, L.; Bao, Y.; Zhu, W.; Hu, Y.; Ma, L.; Wei, S.; Lan, Y. Screening of Potential Genes and Transcription Factors of Postoperative Cognitive Dysfunction via Bioinformatics Methods. Med. Sci. Monit. 2018, 24, 503. [Google Scholar] [CrossRef]
- Santos-Terra, J.; Deckmann, I.; Fontes-Dutra, M.; Schwingel, G.B.; Bambini-Junior, V.; Gottfried, C. Transcription factors in neurodevelopmental and associated psychiatric disorders: A potential convergence for genetic and environmental risk factors. Int. J. Dev. Neurosci. 2021, 81, 545–578. [Google Scholar] [CrossRef]
- Johnston, M.V.; Alemi, L.; Harum, K.H. Learning, Memory, and Transcription Factors. Pediatr. Res. 2003, 53, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.; Schulze-Osthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005, 12 (Suppl. 1), 942–961. [Google Scholar] [CrossRef]
- Razavi, S.; Nazem, G.; Mardani, M.; Esfandiari, E.; Salehi, H.; Esfahani, S.H.Z. Neurotrophic factors and their effects in the treatment of multiple sclerosis. Adv. Biomed. Res. 2015, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, O.; Saarma, M. Neurotrophic Factors in Parkinson’s Disease: Clinical Trials, Open Challenges and Nanoparticle-Mediated Delivery to the Brain. Front. Cell. Neurosci. 2021, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shehadah, A.; Pal, A.; Zacharek, A.; Cui, X.; Cui, Y.; Roberts, C.; Lu, M.; Zeitlin, A.; Hariri, R.; et al. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Padmakumar, S.; Taha, M.S.; Kadakia, E.; Bleier, B.S.; Amiji, M.M. Delivery of neurotrophic factors in the treatment of age-related chronic neurodegenerative diseases. Expert Opin. Drug Deliv. 2020, 17, 323–340. [Google Scholar] [CrossRef]
- Chmielarz, P.; Saarma, M. Neurotrophic factors for disease-modifying treatments of Parkinson’s disease: Gaps between basic science and clinical studies. Pharmacol. Rep. 2020, 72, 1195–1217. [Google Scholar] [CrossRef]
- Alfonsetti, M.; D’Angelo, M.; Castelli, V. Neurotrophic factor-based pharmacological approaches in neurological disorders. Neural Regen. Res. 2023, 18, 1220–1228. [Google Scholar] [CrossRef]
- Reagents for Therapeutic Targets of Neurodegenerative Diseases: Get Quote, RFQ, Price or Buy. Available online: https://www.news-medical.net/Reagents-for-therapeutic-targets-of-neurodegenerative-diseases (accessed on 9 February 2023).
- Saint-André, V. Computational biology approaches for mapping transcriptional regulatory networks. Comput. Struct. Biotechnol. J. 2021, 19, 4884–4895. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.E.; Gonye, G.E.; Schwaber, J.S.; Doyle, F.J. Importance of Input Perturbations and Stochastic Gene Expression in the Reverse Engineering of Genetic Regulatory Networks: Insights from an Identifiability Analysis of an In silico Network. Genome Res. 2003, 13, 2396–2405. [Google Scholar] [CrossRef] [Green Version]
- Zoppoli, P.; Morganella, S.; Ceccarelli, M. TimeDelay-ARACNE: Reverse engineering of gene networks from time-course data by an information theoretic approach. BMC Bioinform. 2010, 11, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Stolovitzky, G.; Monroe, D.; Califano, A. Dialogue on reverse-engineering assessment and methods: The DREAM of high-throughput pathway inference. Ann. N. Y. Acad. Sci. 2007, 1115, 1–22. [Google Scholar] [CrossRef]
- van der Sande, M.; Frölich, S.; van Heeringen, S.J. Computational approaches to understand transcription regulation in development. Biochem. Soc. Trans. 2023, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, J.; Wurtmann, E.J.; Baliga, N.S. Reverse engineering systems models of regulation: Discovery, prediction and mechanisms. Curr. Opin. Biotechnol. 2012, 23, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bastiani, M.A.; Pfaffenseller, B.; Klamt, F. Master regulators connectivity map: A transcription factors-centered approach to drug repositioning. Front. Pharmacol. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carré, C.; Mas, A.; Krouk, G. Reverse engineering highlights potential principles of large gene regulatory network design and learning. NPJ Syst. Biol. Appl. 2017, 3, 17. [Google Scholar] [CrossRef]
- Perkel, J.M. Smart software untangles gene regulation in cells. Nature 2022, 609, 428–431. [Google Scholar] [CrossRef]
- Villaverde, A.F.; Banga, J.R. Reverse engineering and identification in systems biology: Strategies, perspectives and challenges. J. R. Soc. Interface 2014, 11, 20130505. [Google Scholar] [CrossRef] [Green Version]
- Vohradsky, J. Neural model of the genetic network. J. Biol. Chem. 2001, 276, 36168–36173. [Google Scholar] [CrossRef] [Green Version]
- De Cegli, R.; Iacobacci, S.; Flore, G.; Gambardella, G.; Mao, L.; Cutillo, L.; Lauria, M.; Klose, J.; Illingworth, E.; Banfi, S.; et al. Reverse engineering a mouse embryonic stem cell-specific transcriptional network reveals a new modulator of neuronal differentiation. Nucleic Acids Res. 2013, 41, 711–726. [Google Scholar] [CrossRef] [Green Version]
- Acquaah-Mensah, G.K.; Taylor, R.C. Brain in situ hybridization maps as a source for reverse-engineering transcriptional regulatory networks: Alzheimer’s disease insights. Gene 2016, 586, 77–86. [Google Scholar] [CrossRef]
- Petrovskiy, E.D.; Saik, O.V.; Tiys, E.S.; Lavrik, I.N.; Kolchanov, N.A.; Ivanisenko, V.A. Prediction of tissue-specific effects of gene knockout on apoptosis in different anatomical structures of human brain. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Yin, Y.; Zhang, A.; Bernstein, A.M.; Kawaguchi, R.; Gao, K.; Potter, K.; Gilbert, H.Y.; Ao, Y.; Ou, J.; et al. Transcription factor network analysis identifies REST/NRSF as an intrinsic regulator of CNS regeneration in mice. Nat. Commun. 2022, 13, 4418. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rieckhof, G.; Califano, A. Reverse-engineering human regulatory networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Dusonchet, J.; Li, H.; Guillily, M.; Liu, M.; Stafa, K.; Derada Troletti, C.; Boon, J.Y.; Saha, S.; Glauser, L.; Mamais, A.; et al. A Parkinson’s disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum. Mol. Genet. 2014, 23, 4887–4905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łastowska, M.; Al-Afghani, H.; Al-Balool, H.H.; Sheth, H.; Mercer, E.; Coxhead, J.M.; Redfern, C.P.F.; Peters, H.; Burt, A.D.; Santibanez-Koref, M.; et al. Identification of a neuronal transcription factor network involved in medulloblastoma development. Acta Neuropathol. Commun. 2014, 2, 35. [Google Scholar] [CrossRef] [Green Version]
- Fromental-Ramain, C.; Warot, X.; Lakkaraju, S.; Favier, B.; Haack, H.; Birling, C.; Dierich, A.; Dolle, P.; Chambon, P. Specific and redundant functions of the paralogous Hoxa-9 and Hoxd-9 genes in forelimb and axial skeleton patterning. Development 1996, 122, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Domsch, K.; Papagiannouli, F.; Lohmann, I. The HOX-Apoptosis Regulatory Interplay in Development and Disease. Curr. Top. Dev. Biol. 2015, 114, 121–158. [Google Scholar] [CrossRef]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX genes in stem cell differentiation and cancer. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, J.; Wilkinson, D.G. Establishing neuronal circuitry: Hox genes make the connection. Genes Dev. 2004, 18, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, C.S.; Le Boiteux, E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell. Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef]
- Finch, N.A.; Wang, X.; Baker, M.C.; Heckman, M.G.; Gendron, T.F.; Bieniek, K.F.; Wuu, J.; Dejesus-Hernandez, M.; Brown, P.H.; Chew, J.; et al. Abnormal expression of homeobox genes and transthyretin in C9ORF72 expansion carriers. Neurol. Genet. 2017, 3, e161. [Google Scholar] [CrossRef] [Green Version]
- Goodman, F.R.; Majewski, F.; Collins, A.L.; Scambler, P.J. A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am. J. Hum. Genet. 2002, 70, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Sneha, N.P.; Dharshini, S.A.P.; Taguchi, Y.H.; Gromiha, M.M. Integrative Meta-Analysis of Huntington’s Disease Transcriptome Landscape. Genes 2022, 13, 2385. [Google Scholar] [CrossRef]
- Kocak, H.; Ackermann, S.; Hero, B.; Kahlert, Y.; Oberthuer, A.; Juraeva, D.; Roels, F.; Theissen, J.; Westermann, F.; Deubzer, H.; et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013, 4, e586. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Dai, W.; Li, J.; Xiang, L.; Wu, X.; Tang, W.; Chen, Y.; Yang, Q.; Liu, M.; Xiao, Y.; et al. HOXD9 promotes the growth, invasion and metastasis of gastric cancer cells by transcriptional activation of RUFY3. J. Exp. Clin. Cancer Res. 2019, 38, 412. [Google Scholar] [CrossRef] [Green Version]
- Tabuse, M.; Ohta, S.; Ohashi, Y.; Fukaya, R.; Misawa, A.; Yoshida, K.; Kawase, T.; Saya, H.; Thirant, C.; Chneiweiss, H.; et al. Functional analysis of HOXD9 in human gliomas and glioma cancer stem cells. Mol. Cancer 2011, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Kuert, P.A.; Hartenstein, V.; Bello, B.C.; Lovick, J.K.; Reichert, H. Neuroblast lineage identification and lineage-specific Hox gene action during postembryonic development of the subesophageal ganglion in the Drosophila central brain. Dev. Biol. 2014, 390, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Volakakis, N.; Kadkhodaei, B.; Joodmardi, E.; Wallis, K.; Panman, L.; Silvaggi, J.; Spiegelman, B.M.; Perlmann, T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. USA 2010, 107, 12317–12322. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.L.; Yoshida, H.; Yamaguchi, M. Nuclear transcription factor Y and its roles in cellular processes related to human disease. Am. J. Cancer Res. 2013, 3, 339–346. [Google Scholar]
- Herring, J.A.; Elison, W.S.; Tessem, J.S. Function of Nr4a Orphan Nuclear Receptors in Proliferation, Apoptosis and Fuel Utilization Across Tissues. Cells 2019, 8, 1373. [Google Scholar] [CrossRef] [Green Version]
- Hale, T.K.; Myers, C.; Maitra, R.; Kolzau, T.; Nishizawa, M.; Braithwaite, A.W. Maf transcriptionally activates the mouse p53 promoter and causes a p53- dependent cell death. J. Biol. Chem. 2000, 275, 17991–17999. [Google Scholar] [CrossRef] [Green Version]
- Jeanneteau, F.; Barrè, C.; Vos, M.; De Vries, C.J.M.; Rouillard, X.; Levesque, D.; Dromard, Y.; Moisan, M.-P.; Duric, V.; Franklin, T.C.; et al. The Stress-Induced Transcription Factor NR4A1 Adjusts Mitochondrial Function and Synapse Number in Prefrontal Cortex. J. Neurosci. 2018, 38, 1335–1350. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, Y.; Ertürk, A.; Kallop, D.; Jiang, Z.; Weimer, R.M.; Kaminker, J.; Sheng, M. Activity-induced Nr4a1 regulates spine density and distribution pattern of excitatory synapses in pyramidal neurons. Neuron 2014, 83, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Jakaria, M.; Haque, M.E.; Cho, D.-Y.; Azam, S.; Kim, I.-S.; Choi, D.-K. Molecular Insights into NR4A2(Nurr1): An Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death. Mol. Neurobiol. 2019, 56, 5799–5814. [Google Scholar] [CrossRef]
- Corley, S.M.; Tsai, S.Y.; Wilkins, M.R.; Weickert, C.S. Transcriptomic analysis shows decreased cortical expression of nr4a1, nr4a2 and rxrb in schizophrenia and provides evidence for nuclear receptor dysregulation. PLoS ONE 2016, 11, e0166944. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.-Y.; Catts, V.S.; Fullerton, J.M.; Corley, S.M.; Fillman, S.G.; Weickert, C.S. Nuclear Receptors and Neuroinflammation in Schizophrenia. Mol. Neuropsychiatry 2017, 3, 181–191. [Google Scholar] [CrossRef]
- Xiao, G.; Sun, T.; Songming, C.; Cao, Y. NR4A1 enhances neural survival following oxygen and glucose deprivation: An in vitro study. J. Neurol. Sci. 2013, 330, 78–84. [Google Scholar] [CrossRef]
- Rouillard, C.; Baillargeon, J.; Paquet, B.; St-Hilaire, M.; Maheux, J.; Lévesque, C.; Darlix, N.; Majeur, S.; Lévesque, D. Genetic disruption of the nuclear receptor Nur77 (Nr4a1) in rat reduces dopamine cell loss and L-Dopa-induced dyskinesia in experimental Parkinson’s disease. Exp. Neurol. 2018, 304, 143–153. [Google Scholar] [CrossRef]
- Zhao, L.G.; Tang, Y.; Tan, J.Z.; Wang, J.W.; Chen, G.J.; Zhu, B.L. The effect of NR4A1 on APP metabolism and tau phosphorylation. Genes Dis. 2018, 5, 342–348. [Google Scholar] [CrossRef]
- Bao, X.J.; Wang, G.C.; Zuo, F.X.; Li, X.Y.; Wu, J.; Chen, G.; Dou, W.C.; Guo, Y.; Shen, Q.; Wang, R.Z. Transcriptome profiling of the subventricular zone and dentate gyrus in an animal model of Parkinson’s disease. Int. J. Mol. Med. 2017, 40, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Walsh, E.N.; Yan, A.L.; Giese, K.P.; Safe, S.; Abel, T. Pharmacological activation of Nr4a rescues age-associated memory decline. Neurobiol. Aging 2020, 85, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Tello, P.; Lin, H.; Khan, P.; de Vera, I.M.; Kamenecka, T.; Kojetin, D. Assessment of NR4A Ligands that Directly Bind and Modulate the Orphan Nuclear Receptor Nurr1. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Bridi, M.S.; Hawk, J.D.; Chatterjee, S.; Safe, S.; Abel, T. Pharmacological Activators of the NR4A Nuclear Receptors Enhance LTP in a CREB/CBP-Dependent Manner. Nat. Publ. Gr. 2016, 42, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Sánchez, E.; Jiménez-Genchi, J.; Alcántara-Flores, Y.M.; Castañeda-González, C.J.; Aviña-Cervantes, C.L.; Yescas, P.; del Socorro González-Valadez, M.; Martínez-Rodríguez, N.; Ríos-Ortiz, A.; González-González, M.; et al. Working memory deficits in schizophrenia are associated with the rs34884856 variant and expression levels of the NR4A2 gene in a sample Mexican population: A case control study. BMC Psychiatry 2021, 21, 86. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Zai, C.C.; Mirkhani, M.; Shaikh, S.; Vincent, J.B.; Meltzer, H.; Lieberman, J.A.; Strauss, J.; Lévesque, D.; Kennedy, J.L.; et al. Replicated association of the NR4A3 gene with smoking behaviour in schizophrenia and in bipolar disorder. Genes Brain Behav. 2010, 9, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Tashiro, S.; Tsuchiya, H.; Kume, A.; Kanno, M.; Ito, E.; Yamamoto, M.; Igarashi, K. Activation of Maf/AP-1 repressor Bach2 by oxidative stress promotes apoptosis and its interaction with promyelocytic leukemia nuclear bodies. J. Biol. Chem. 2002, 277, 20724–20733. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Lalani, S.; Leavenworth, J.W.; Ho, I.C.; Pauza, M.E. c-Maf interacts with c-Myb to down-regulate Bcl-2 expression and increase apoptosis in peripheral CD4 cells. Eur. J. Immunol. 2007, 37, 2868–2880. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Tamagawa, Y.; Kure, S.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Small Maf compound mutants display central nervous system neuronal degeneration, aberrant transcription, and Bach protein mislocalization coincident with myoclonus and abnormal startle response. Mol. Cell. Biol. 2003, 23, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Mao, H.; He, G.; Mao, X. Targeting the oncogenic transcription factor c-Maf for the treatment of multiple myeloma. Cancer Lett. 2022, 543, 215791. [Google Scholar] [CrossRef]
- Balamurugan, K.; Sterneck, E. The Many Faces of C/EBPδ and their Relevance for Inflammation and Cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef] [Green Version]
- Pulido-Salgado, M.; Vidal-Taboada, J.M.; Saura, J. C/EBPβ and C/EBPδ transcription factors: Basic biology and roles in the CNS. Prog. Neurobiol. 2015, 132, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Kapatos, G. Identification of neuronal target genes for CCAAT/Enhancer Binding Proteins. Mol. Cell. Neurosci. 2009, 40, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Canteli, M.; Aguilar-Morante, D.; Sanz-SanCristobal, M.; Megias, D.; Santos, A.; Perez-Castillo, A. Role of C/EBPβ transcription factor in adult hippocampal neurogenesis. PLoS ONE 2011, 6, e24842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meir, O.; Dvash, E.; Werman, A.; Rubinstein, M. C/EBP-β regulates endoplasmic reticulum stress-triggered cell death in mouse and human models. PLoS ONE 2010, 5, e9516. [Google Scholar] [CrossRef]
- Moore, F.; Santin, I.; Nogueira, T.C.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. The transcription factor C/EBP delta has anti-apoptotic and anti-inflammatory roles in pancreatic beta cells. PLoS ONE 2012, 7, e31062. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Canteli, M.; Luna-Medina, R.; Sanz-SanCristobal, M.; Alvarez-Barrientos, A.; Santos, A.; Perez-Castillo, A. CCAAT/enhancer binding protein β deficiency provides cerebral protection following excitotoxic injury. J. Cell Sci. 2008, 121, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.H.; Gong, K.; Liu, X.; Zhang, Z.; Sun, X.; Wei, Z.Z.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Johnson, P.F.; et al. C/EBPβ regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat. Commun. 2018, 9, 1784. [Google Scholar] [CrossRef] [Green Version]
- Ramberg, V.; Tracy, L.M.; Samuelsson, M.; Nilsson, L.N.G.; Iverfeldt, K. The CCAAT/enhancer binding protein (C/EBP) δ is differently regulated by fibrillar and oligomeric forms of the Alzheimer amyloid-β peptide. J. Neuroinflamm. 2011, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, S.; Hein, P.; Fernandes, K.J.L.; Peterson, A.C.; Miller, F.D. A transcriptional role for C/EBP β in the neuronal response to axonal injury. Mol. Cell. Neurosci. 2005, 29, 525–535. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, C.T.; Chiang, C.K.; Liao, B.W.; Liu, S.H. C/EBP Homologous Protein (CHOP) Deficiency Aggravates Hippocampal Cell Apoptosis and Impairs Memory Performance. PLoS ONE 2012, 7, e40801. [Google Scholar] [CrossRef]
- Nashine, S.; Liu, Y.; Kim, B.J.; Clark, A.F.; Pang, I.H. Role of C/EBP Homologous Protein in Retinal Ganglion Cell Death After Ischemia/Reperfusion Injury. Invest. Ophthalmol. Vis. Sci. 2015, 56, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, Y.; Ishiwata, S.; Hoshino, M. Characterization of Olig2 expression during cerebellar development. Gene Expr. Patterns 2014, 15, 1–7. [Google Scholar] [CrossRef]
- Gaber, Z.B.; Novitch, B.G. All the embryo’s a stage, and Olig2 in its time plays many parts. Neuron 2011, 69, 833–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esain, V.; Postlethwait, J.H.; Charnay, P.; Ghislain, J. FGF-receptor signalling controls neural cell diversity in the zebrafish hindbrain by regulating olig2 and sox9. Development 2010, 137, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.L.; Fragoso, G.; Miron, V.E.; Darlington, P.J.; Mushynski, W.E.; Antel, J.; Almazan, G. Response of human oligodendrocyte progenitors to growth factors and axon signals. J. Neuropathol. Exp. Neurol. 2010, 69, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Allahdadi, K.J.; De Santana, T.A.; Santos, G.C.; Azevedo, C.M.H.; Mota, R.A.; Nonaka, C.K.; Silva, D.N.; Valim, C.X.R.; Figueira, C.P.; Dos Santos, W.L.C.; et al. IGF-1 overexpression improves mesenchymal stem cell survival and promotes neurological recovery after spinal cord injury. Stem Cell Res. Ther. 2019, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Furusho, M.; Kaga, Y.; Ishii, A.; Hébert, J.M.; Bansal, R. Fibroblast Growth Factor Signaling Is Required for the Generation of Oligodendrocyte Progenitors from the Embryonic Forebrain. J. Neurosci. 2011, 31, 5055–5066. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.; Hollingworth, P.; Moskvina, V.; Dowzell, K.; O’Donovan, M.C.; Powell, J.; Lovestone, S.; Brayne, C.; Rubinsztein, D.; Owen, M.J.; et al. Evidence that variation in the oligodendrocyte lineage transcription factor 2 (OLIG2) gene is associated with psychosis in Alzheimer’s disease. Neurosci. Lett. 2009, 461, 54–59. [Google Scholar] [CrossRef]
- Komatsu, H.; Takeuchi, H.; Kikuchi, Y.; Ono, C.; Yu, Z.; Iizuka, K.; Takano, Y.; Kakuto, Y.; Funakoshi, S.; Ono, T.; et al. Ethnicity-Dependent Effects of Schizophrenia Risk Variants of the OLIG2 Gene on OLIG2 Transcription and White Matter Integrity. Schizophr. Bull. 2020, 46, 1619–1628. [Google Scholar] [CrossRef]
- Gouvêa-Junqueira, D.; Falvella, A.C.B.; Antunes, A.S.L.M.; Seabra, G.; Brandão-Teles, C.; Martins-de-Souza, D.; Crunfli, F. Novel Treatment Strategies Targeting Myelin and Oligodendrocyte Dysfunction in Schizophrenia. Front. Psychiatry 2020, 11, 379. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Mukthavaram, R.; Kouznetsova, V.L.; Chao, Y.; Babic, I.; Nurmemmedov, E.; Pastorino, S.; Jiang, P.; Calligaris, D.; Agar, N.; et al. Multiple spatially related pharmacophores define small molecule inhibitors of OLIG2 in glioblastoma. Oncotarget 2015, 8, 22370–22384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, G.; Kesari, S. Novel small molecule inhibitors of the OLIG2 transcription factor: Promising new therapeutics for glioblastoma. Future Oncol. 2016, 12, 1001–1004. [Google Scholar] [CrossRef]
- Chen, X.; Wang, F.; Gan, J.; Zhang, Z.; Liang, X.; Li, T.; Huang, N.; Zhao, X.; Mei, F.; Xiao, L. Myelin Deficits Caused by Olig2 Deficiency Lead to Cognitive Dysfunction and Increase Vulnerability to Social Withdrawal in Adult Mice. Neurosci. Bull. 2020, 36, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.; Greenall, S.; Chen, S.; Stewart, R.; Alton, G.; Kesari, S. DDIS-19. CT-179: An Inhibitor of The Olig2 Transcription Factor with Potent Anti-Tumour Activity In Brain Cancer. Neuro. Oncol. 2018, 20, vi73. [Google Scholar] [CrossRef]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain 2015, 138, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Szu, J.; Wojcinski, A.; Jiang, P.; Kesari, S. Impact of the Olig Family on Neurodevelopmental Disorders. Front. Neurosci. 2021, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y. Molecular mechanisms of regeneration in Alzheimer’s disease brain. Geriatr. Gerontol. Int. 2010, 10, S158–S168. [Google Scholar] [CrossRef]
- Stewart, S.E.; Platko, J.; Fagerness, J.; Birns, J.; Jenike, E.; Smoller, J.W.; Perlis, R.; Leboyer, M.; Delorme, R.; Chabane, N.; et al. A genetic family-based association study of OLIG2 in obsessive-compulsive disorder. Arch. Gen. Psychiatry 2007, 64, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.-I.; Asahina, N.; Kitano, S.; Kino, Y. A Comprehensive Profile of ChIP-Seq-Based Olig2 Target Genes in Motor Neuron Progenitor Cells Suggests the Possible Involvement of Olig2 in the Pathogenesis of Amyotrophic Lateral Sclerosis. J. Cent. Nerv. Syst. Dis. 2015, 7, JCNSD.S23210. [Google Scholar] [CrossRef]
- Tan, B.T.; Yu, J.; Yin, Y.; Jia, G.W.; Jiang, W.; Yu, L.H. The Olig family affects central nervous system development and disease. Neural Regen. Res. 2014, 9, 329–336. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, Z.; Ransom, B.R.; Tong, X. Oligodendrocyte lineage cells and depression. Mol. Psychiatry 2020, 26, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Raabe, F.J.; Galinski, S.; Papiol, S.; Falkai, P.G.; Schmitt, A.; Rossner, M.J. Studying and modulating schizophrenia-associated dysfunctions of oligodendrocytes with patient-specific cell systems. NPJ Schizophr. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessel, J. Formulating treatment of major psychiatric disorders: Algorithm targets the dominantly affected brain cell-types. Discov. Ment. Health 2023, 3, 3. [Google Scholar] [CrossRef]
- Cai, S.; Lv, Y.; Huang, K.; Zhang, W.; Kang, Y.; Huang, L.; Wang, J. Association of rs1059004 polymorphism in the OLIG2 locus with whole-brain functional connectivity in first-episode schizophrenia. Behav. Brain Res. 2020, 379, 112392. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tovar, M.; Rodríguez-Ramírez, A.M.; Rodríguez-Cárdenas, L.; Sotelo-Ramírez, C.E.; Camarena, B.; Sanabrais-Jiménez, M.A.; Solís-Chagoyán, H.; Argueta, J.; López-Riquelme, G.O. Insights into myelin dysfunction in schizophrenia and bipolar disorder. World J. Psychiatry 2022, 12, 264. [Google Scholar] [CrossRef]
- Van Der Raadt, J.; Van Gestel, S.H.C.; Kasri, N.N.; Albers, C.A. ONECUT transcription factors induce neuronal characteristics and remodel chromatin accessibility. Nucleic Acids Res. 2019, 47, 5587–5602. [Google Scholar] [CrossRef]
- Patel, T.; Hammelman, J.; Closser, M.; Gifford, D.K.; Wichterle, H. General and cell-type-specific aspects of the motor neuron maturation transcriptional program. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hahn, O.; Foltz, A.G.; Atkins, M.; Kedir, B.; Moran-Losada, P.; Guldner, I.H.; Munson, C.; Kern, F.; Pálovics, R.; Lu, N.; et al. A spatiotemporal map of the aging mouse brain reveals white matter tracts as vulnerable foci. bioRxiv 2022. [Google Scholar] [CrossRef]
- Chakrabarty, K.; Von Oerthel, L.; Hellemons, A.; Clotman, F.; Espana, A.; Koerkamp, M.G.; Holstege, F.C.P.; Pasterkamp, R.J.; Smidt, M.P. Genome wide expression profiling of the mesodiencephalic region identifies novel factors involved in early and late dopaminergic development. Biol. Open 2012, 1, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Toch, M.; Harris, A.; Schakman, O.; Kondratskaya, E.; Boulland, J.L.; Dauguet, N.; Debrulle, S.; Baudouin, C.; Hidalgo-Figueroa, M.; Gow, A.; et al. Onecut-dependent Nkx6.2 transcription factor expression is required for proper formation and activity of spinal locomotor circuits. Sci. Rep. 2020, 10, 996. [Google Scholar] [CrossRef] [Green Version]
- Ulmke, P.A.; Sakib, M.S.; Ditte, P.; Sokpor, G.; Kerimoglu, C.; Pham, L.; Xie, Y.; Mao, X.; Rosenbusch, J.; Teichmann, U.; et al. Molecular Profiling Reveals Involvement of ESCO2 in Intermediate Progenitor Cell Maintenance in the Developing Mouse Cortex. Stem Cell Rep. 2021, 16, 968–984. [Google Scholar] [CrossRef]
- Francius, C.; Clotman, F. Dynamic expression of the Onecut transcription factors HNF-6, OC-2 and OC-3 during spinal motor neuron development. Neuroscience 2010, 165, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hin, N.; Newman, M.; Kaslin, J.; Douek, A.M.; Lumsden, A.; Nik, S.H.M.; Dong, Y.; Zhou, X.F.; Manucat-Tan, N.B.; Ludington, A.; et al. Accelerated brain aging towards transcriptional inversion in a zebrafish model of the K115fs mutation of human PSEN2. PLoS ONE 2020, 15, e0227258. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Wang, M.; Cui, Y.; Tan, Z.; Jiang, Y. Differential Expression Profile of lncRNA in Glioma Cells and the Effect of lncRNA NKX3-1 on Glioma Cells Through Fem1b/SPDEF Pathway. Front. Oncol. 2021, 11, 2734. [Google Scholar] [CrossRef] [PubMed]
- Ducker, C.; Shaw, P.E. Ubiquitin-Mediated Control of ETS Transcription Factors: Roles in Cancer and Development. Int. J. Mol. Sci. 2021, 22, 5119. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Mou, X.; Deng, J.; Di, B.; Zhong, R.; Wang, S.; Yang, Y.; Zeng, W. Differences of immune disorders between Alzheimer’s disease and breast cancer based on transcriptional regulation. PLoS ONE 2017, 12, e0180337. [Google Scholar] [CrossRef] [Green Version]
- Tansey, K.E.; Cameron, D.; Hill, M.J. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Arefin, A.S.; Mathieson, L.; Johnstone, D.; Berretta, R.; Moscato, P. Unveiling Clusters of RNA Transcript Pairs Associated with Markers of Alzheimer’s Disease Progression. PLoS ONE 2012, 7, e45535. [Google Scholar] [CrossRef]
- Tay, N.; Macare, C.; Liu, Y.; Ruggeri, B.; Jia, T.; Chu, C.; Biondo, F.; Ing, A.; Luo, Q.; Sarkysian, D.; et al. Allele-specific methylation of SpDEF: A novel moderator of psychosocial stress and substance abuse. Am. J. Psychiatry 2019, 176, 146–155. [Google Scholar] [CrossRef]
- Hughes, R.; Kristiansen, M.; Lassot, I.; Desagher, S.; Mantovani, R.; Ham, J. NF-Y is essential for expression of the proapoptotic bim gene in sympathetic neurons. Cell Death Differ. 2010, 18, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, R.; Dolfini, D.; Mantovani, R. NF-Y joins E2Fs, p53 and other stress transcription factors at the apoptosis table. Cell Death Dis. 2011, 2, e162. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Fuschi, P.; Martelli, F.; Manni, I.; Artuso, S.; Simonte, G.; Ambrosino, V.; Antonini, A.; Folgiero, V.; Falcioni, R.; et al. Transcription factor NF-Y induces apoptosis in cells expressing wild-type p53 through E2F1 upregulation and p53 activation. Cancer Res. 2010, 70, 9711–9720. [Google Scholar] [CrossRef] [Green Version]
- Olajide, O.J.; Suvanto, M.E.; Chapman, C.A. Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer’s disease. Biol. Open 2021, 10, bio056796. [Google Scholar] [CrossRef]
- Yamanaka, T.; Tosaki, A.; Kurosawa, M.; Matsumoto, G.; Koike, M.; Uchiyama, Y.; Maity, S.N.; Shimogori, T.; Hattori, N.; Nukina, N. NF-Y inactivation causes atypical neurodegeneration characterized by ubiquitin and p62 accumulation and endoplasmic reticulum disorganization. Nat. Commun. 2014, 5, 3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or separate entities?—Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Twist Is a Potential Oncogene That Inhibits Apoptosis. Available online: http://genesdev.cshlp.org/content/13/17/2207 (accessed on 9 February 2023).
- Puisieux, A.; Valsesia-Wittmann, S.; Ansieau, S. A twist for survival and cancer progression. Br. J. Cancer 2005, 94, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floc’h, N.; Kolodziejski, J.; Akkari, L.; Simonin, Y.; Ansieau, S.; Puisieux, A.; Hibner, U.; Lassus, P. Modulation of Oxidative Stress by Twist Oncoproteins. PLoS ONE 2013, 8, e72490. [Google Scholar] [CrossRef] [Green Version]
- Jembrek, M.J.; Oršolić, N.; Mandić, L.; Sadžak, A.; Šegota, S. Anti-Oxidative, Anti-Inflammatory and Anti-Apoptotic Effects of Flavonols: Targeting Nrf2, NF-κB and p53 Pathways in Neurodegeneration. Antioxidants 2021, 10, 1628. [Google Scholar] [CrossRef]
- Pan, Y.; Zhu, Y.; Yang, W.; Tycksen, E.; Liu, S.; Palucki, J.; Zhu, L.; Sasaki, Y.; Sharma, M.K.; Kim, A.H.; et al. The role of Twist1 in mutant huntingtin-induced transcriptional alterations and neurotoxicity. J. Biol. Chem. 2018, 293, 11850–11866. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xiang, Y.; Liao, M.J.; Wu, P.F.; Yang, L.; Huang, G.H.; Shi, B.Z.; Yi, L.; Lv, S.Q. Presenilin1 inhibits glioblastoma cell invasiveness via promoting Sortilin cleavage. Cell Commun. Signal. 2021, 19, 1112. [Google Scholar] [CrossRef] [PubMed]

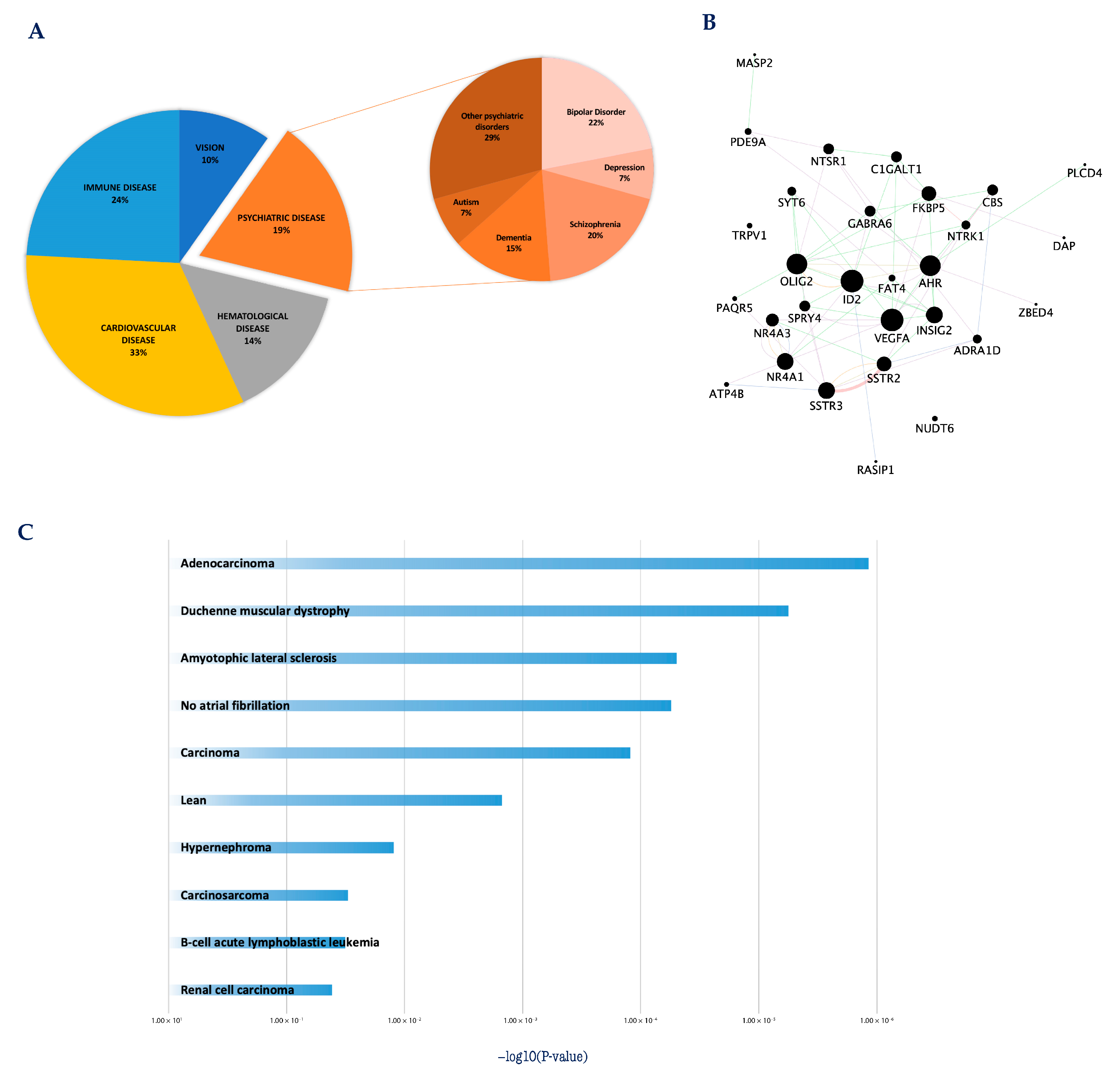
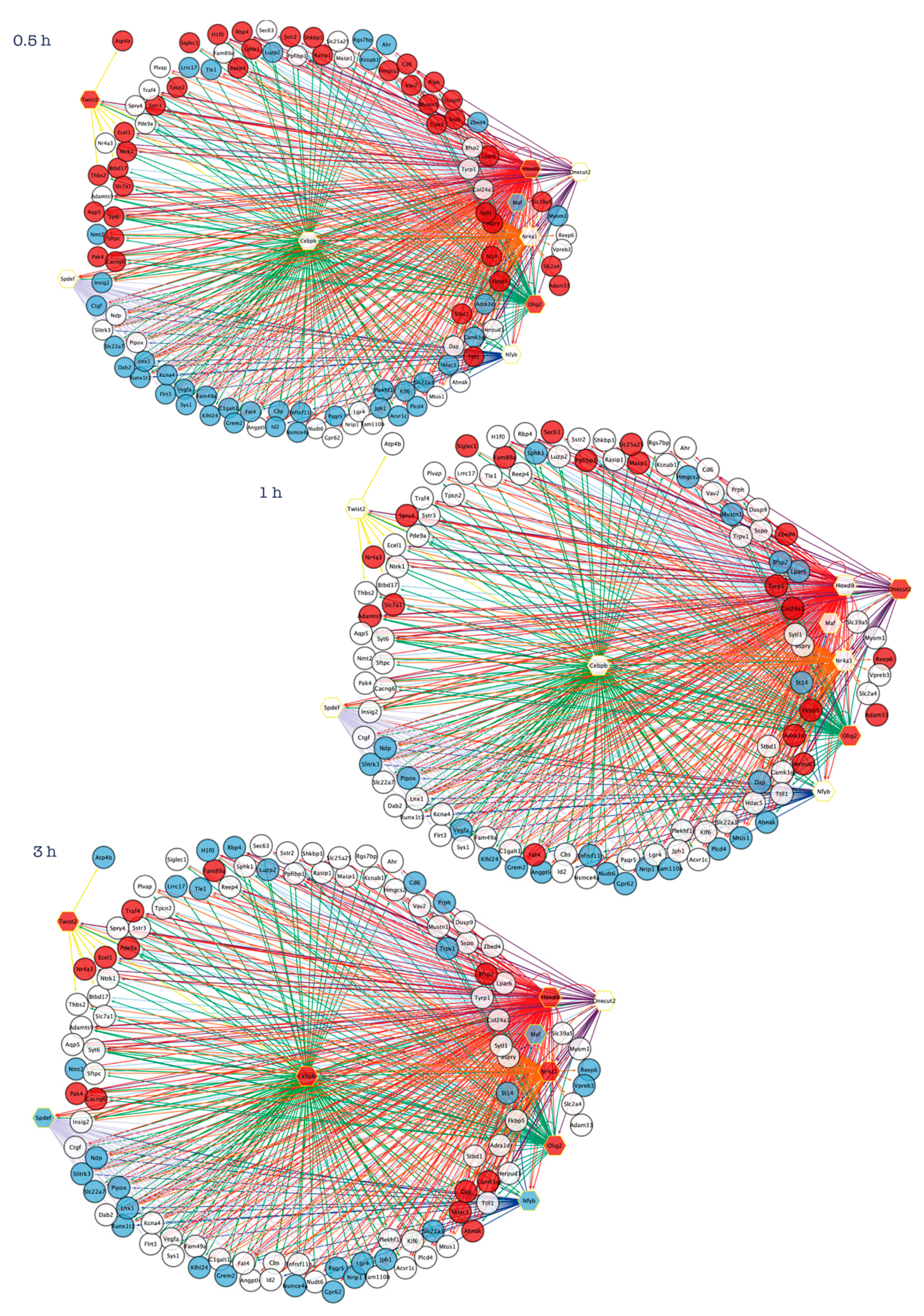

{kind=link}
{kind=link}
{kind=link}
| Perturbation | p-Value | Perturbation | p-Value | |
|---|---|---|---|---|
| Inflammation & Immunologic disorders | Indomethacin | 3.10 × 10−5 | Nystatin | 3.24 × 10−5 |
| Dipyrone | 1.07 × 10−4 | Tranilast | 3.41 × 10−5 | |
| Sulfanilamide | 1.12 × 10−4 | Cyproheptadine | 1.62 × 10−2 | |
| Rifabutin | 2.34 × 10−4 | Rapamycin | 1.66 × 10−6 | |
| Allopurinol | 8.01 × 10−6 | Tacrolimus | 5.22 × 10−5 | |
| Necrostatin | 5.29 × 10−5 | Theophylline | 3.80 × 10−5 | |
| Cancer | Tozasertib | 2.08 × 10−5 | Tyrphostin | 1.54 × 10−15 |
| L-Sulforaphane | 1.00 × 10−4 | Tanespimycin | 7.69 × 10−5 | |
| Psychiatric disorders | Tianeptine | 8.71 × 10−5 | Moclobemide | 2.22 × 10−4 |
| Amitriptyline | 7.97 × 10−6 | Rolipram | 2.15 × 10−4 | |
| Nortriptyline | 8.62 × 10−6 | Azacyclonol | 1.55 × 10−2 | |
| Bupropion | 1.57 × 10−5 | Piracetam | 4.11 × 10−5 | |
| Roflumilast | 1.84 × 10−5 | Promazine hydrochloride | 5.32 × 10−4 | |
| Citalopram | 3.85 × 10−5 | Phenotiazine | 1.09 × 10−4 | |
| Iproniazid | 7.33 × 10−5 | Clozapine | 1.35 × 10−4 | |
| Doxepin | 1.94 × 10−4 | Diazepam | 8.10 × 10−5 | |
| Epilepsy | Lamotrigine | 1.68 × 10−4 | Ethosuximide | 1.07 × 10−4 |
| Cardiovascular diseases | Enaplapril | 2.75 × 10−4 | Atorvastatin | 1.03 × 10−6 |
| Nifedipine | 2.79 × 10−4 | Nicergoline | 1.91 × 10−4 | |
| Other | Monorden/Radicicol | 6.94 × 10−3 | 5-Nonyloxytryptamine | 2.72 × 10−4 |
| Purmorphamine | 1.80 × 10−4 | Parthenolide | 4.11 × 10−5 | |
| Pifithrin | 2.95 × 10−30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morello, G.; La Cognata, V.; Guarnaccia, M.; D’Agata, V.; Cavallaro, S. Cracking the Code of Neuronal Cell Fate. Cells 2023, 12, 1057. https://doi.org/10.3390/cells12071057
Morello G, La Cognata V, Guarnaccia M, D’Agata V, Cavallaro S. Cracking the Code of Neuronal Cell Fate. Cells. 2023; 12(7):1057. https://doi.org/10.3390/cells12071057
Chicago/Turabian StyleMorello, Giovanna, Valentina La Cognata, Maria Guarnaccia, Velia D’Agata, and Sebastiano Cavallaro. 2023. "Cracking the Code of Neuronal Cell Fate" Cells 12, no. 7: 1057. https://doi.org/10.3390/cells12071057
APA StyleMorello, G., La Cognata, V., Guarnaccia, M., D’Agata, V., & Cavallaro, S. (2023). Cracking the Code of Neuronal Cell Fate. Cells, 12(7), 1057. https://doi.org/10.3390/cells12071057