

Development of Cyclic Peptides Targeting the Epidermal Growth Factor Receptor in Mesenchymal Triple-Negative Breast Cancer Subtype

 ,
, 
 , , , ,
, , , ,  , ,
, ,  ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
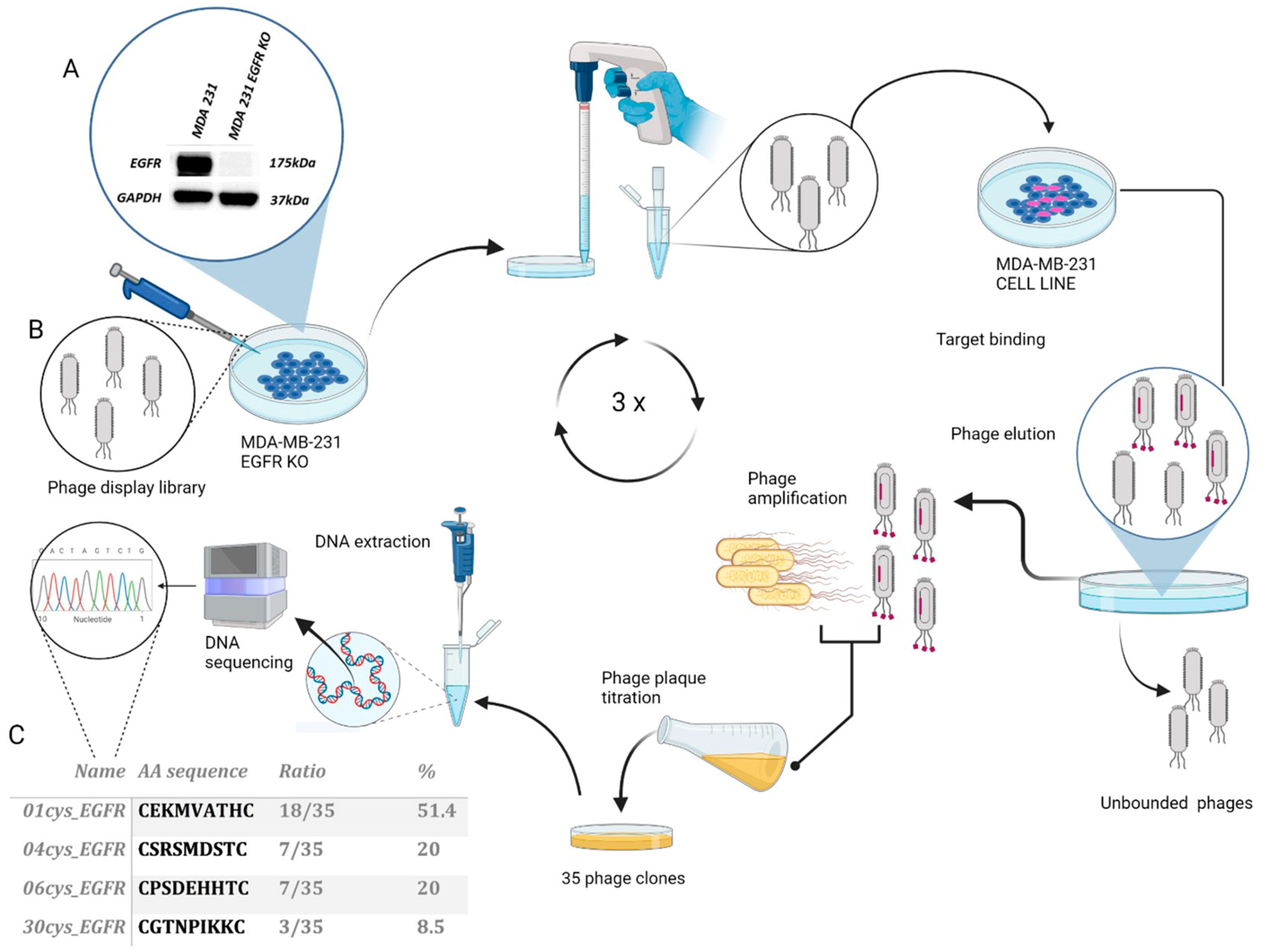
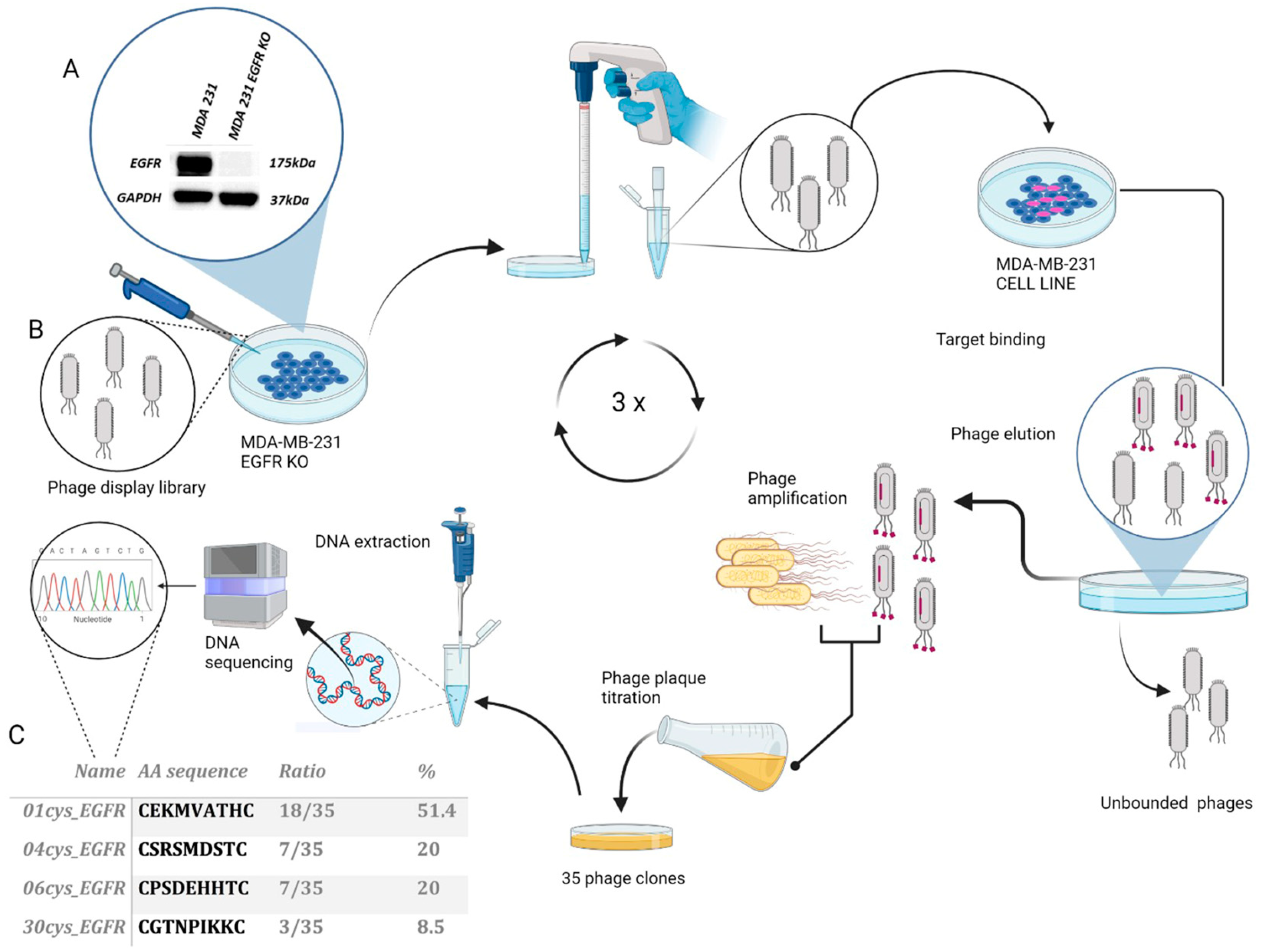
2.2. Phage Display Screening
2.3. Phage DNA Purification and Sequencing
2.4. FITC Phages Labeling Kit
2.5. Bioinformatics Analysis of Peptide Conformation
2.6. Peptides-EGFR Docking Studies
2.7. Peptide Synthesis
2.8. Western Blotting
2.9. Cell Binding Assay
2.10. Cell Cycle Analysis
2.11. Immunofluorescence
2.12. Statistical Analysis
3. Results
3.1. Selection and Identification of EGFR Binding Peptides by Phage Display
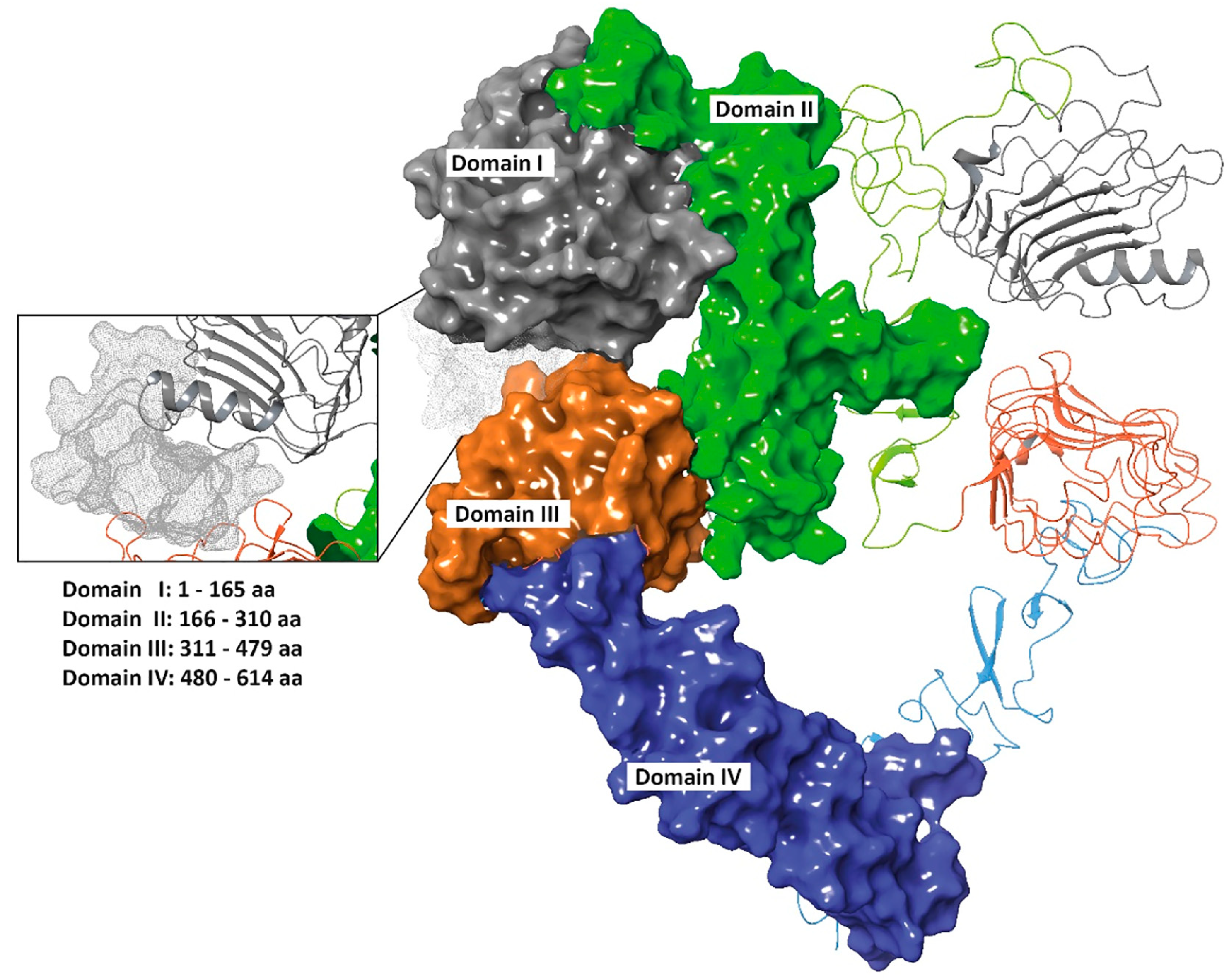
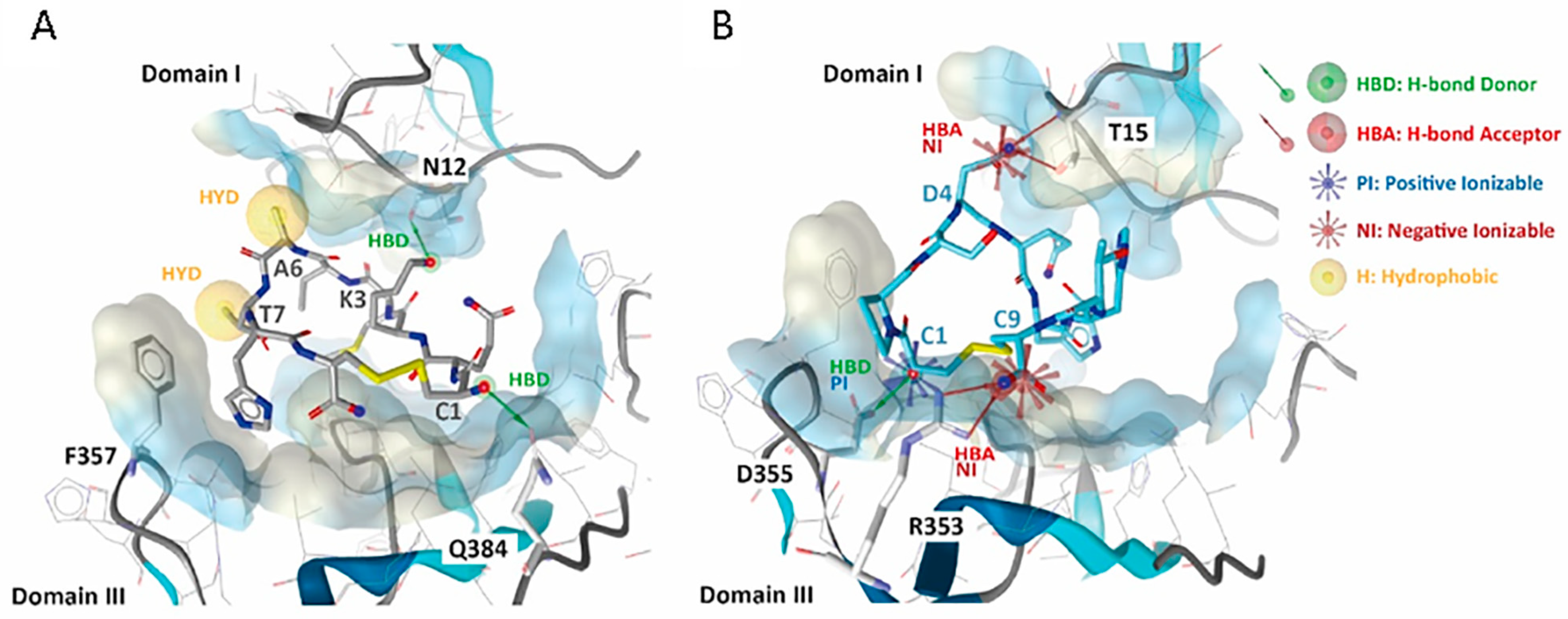
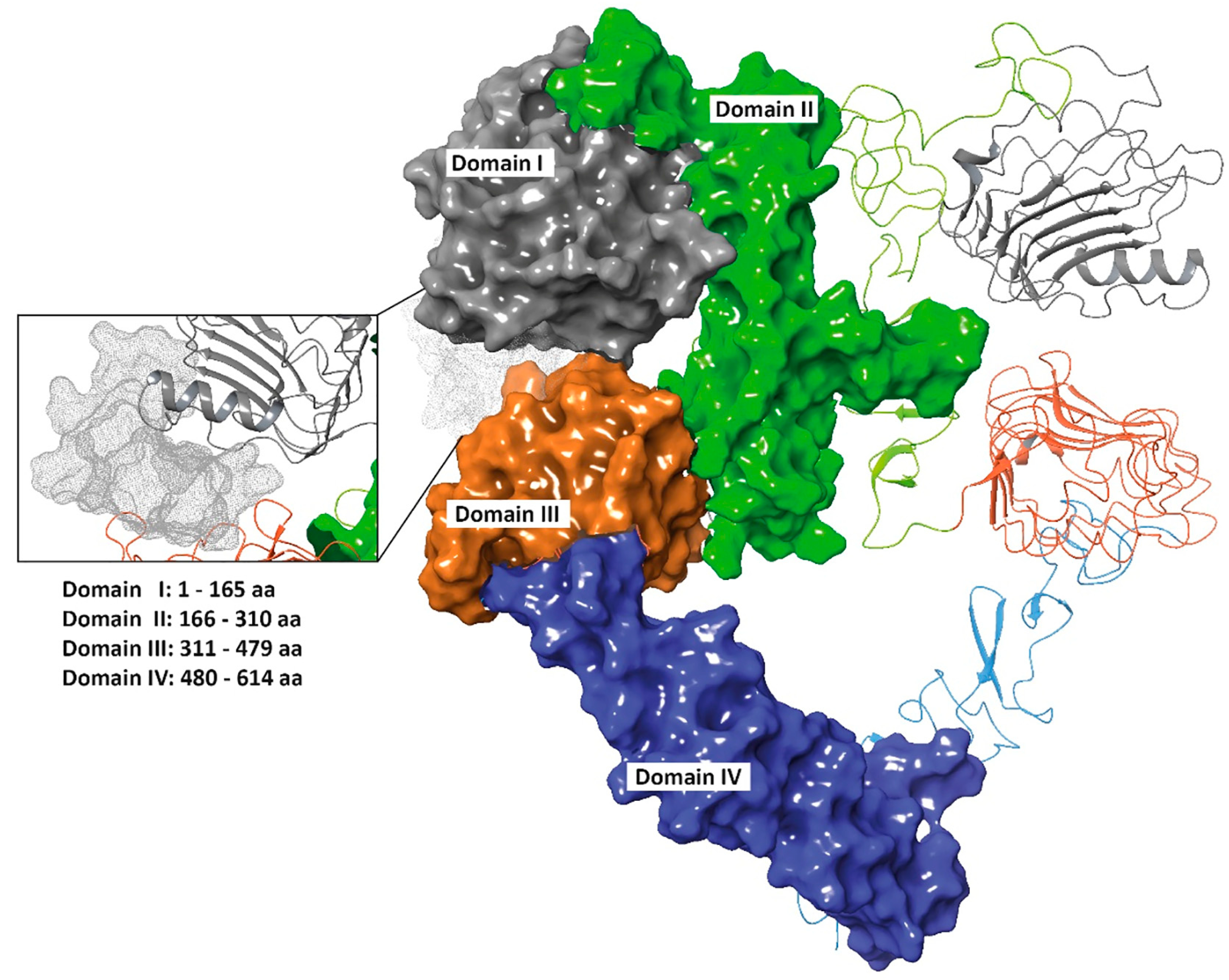
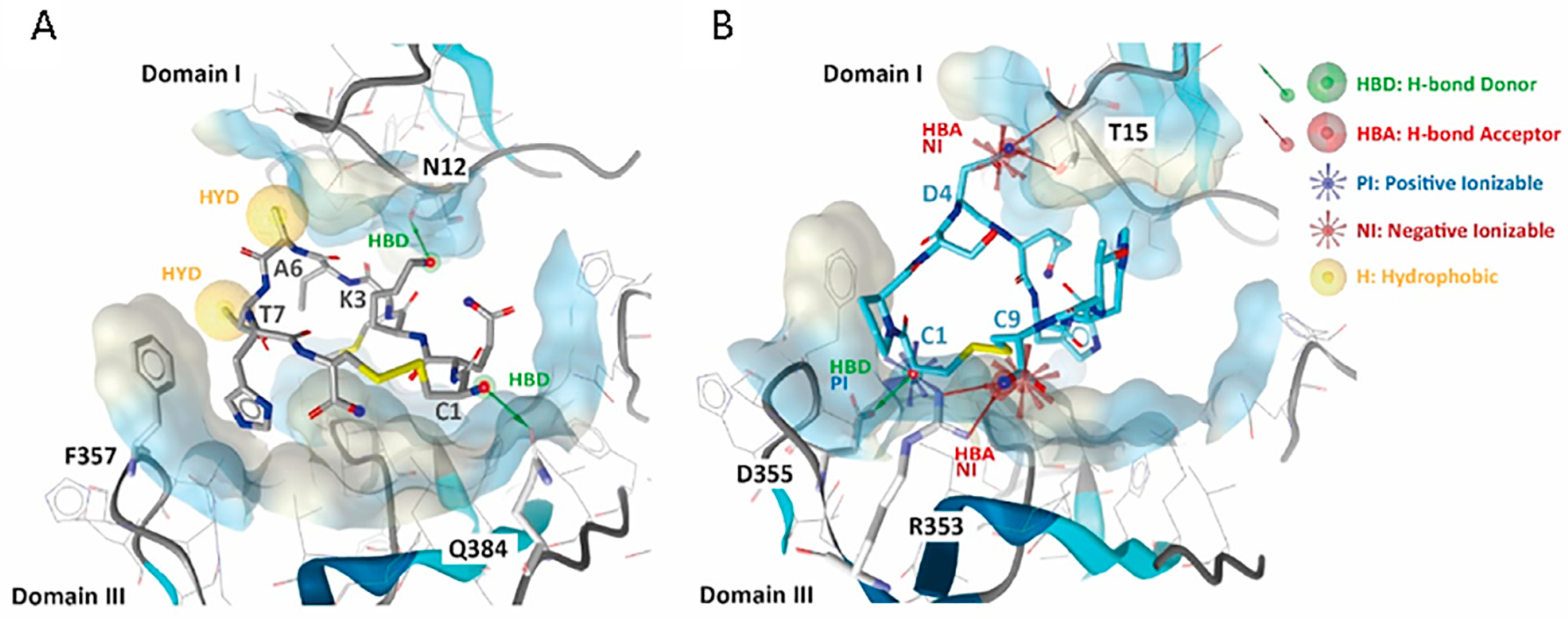
3.2. 01_cys EGFR and 06_cys EGFR Docking Studies in the EGFR Pocket
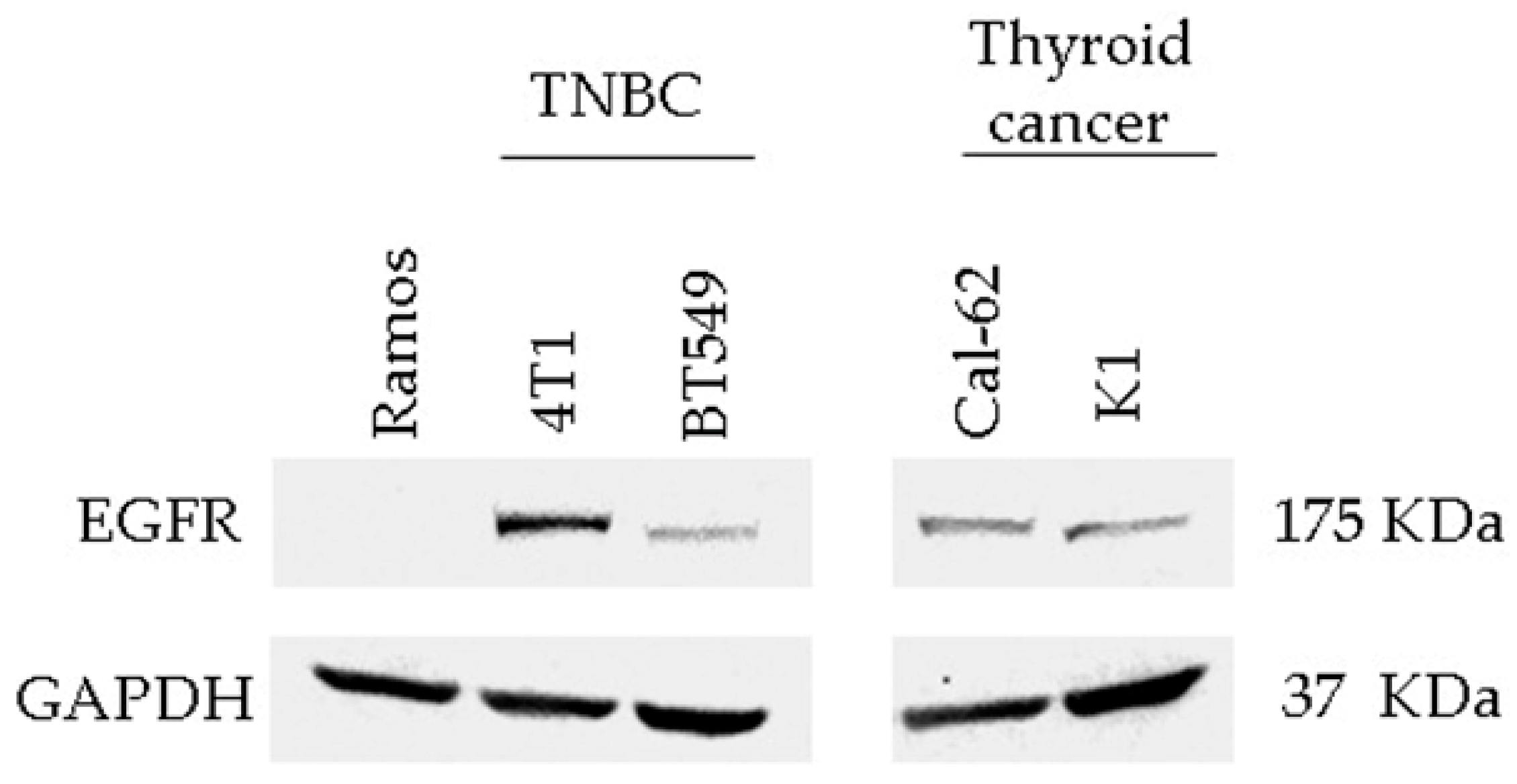
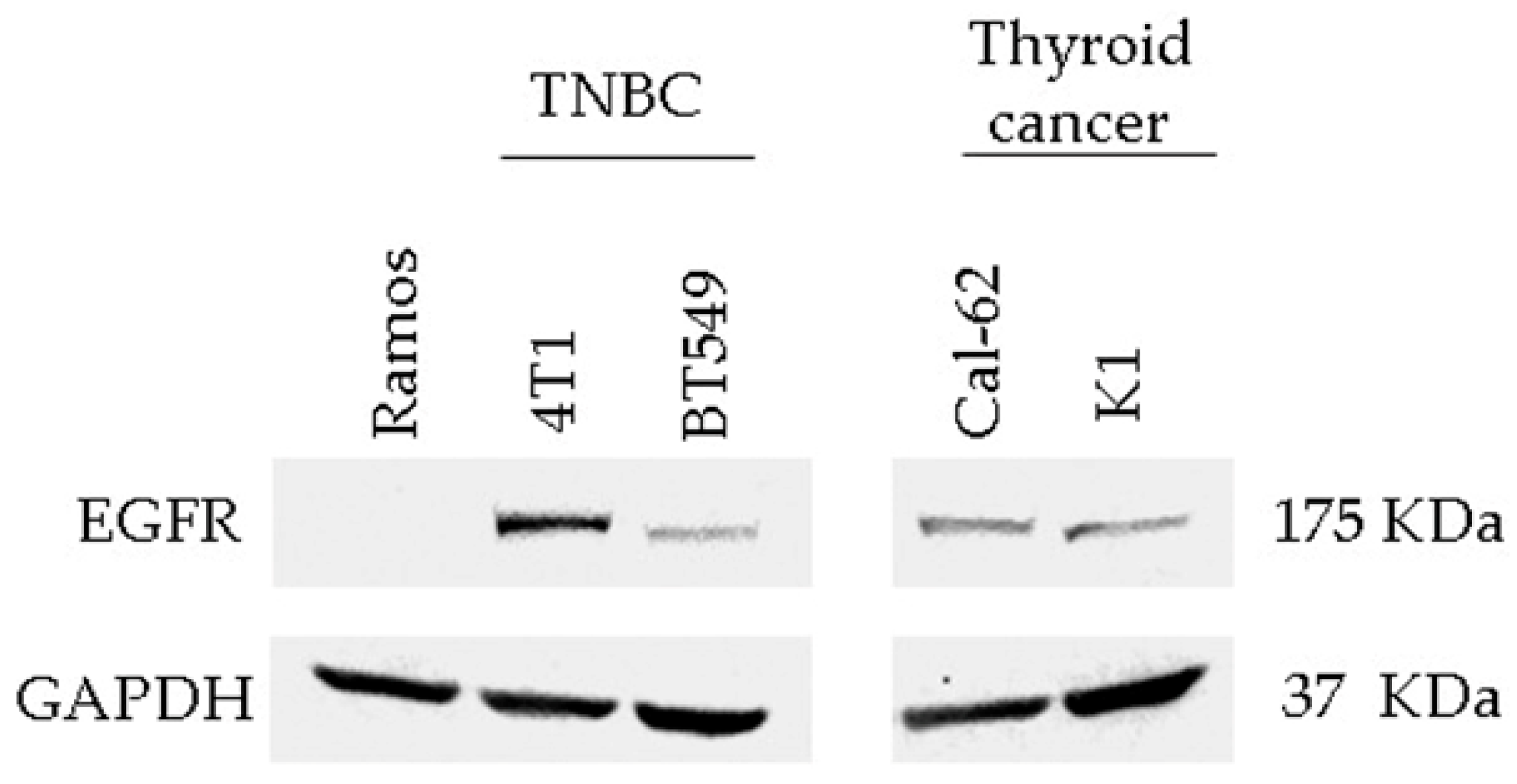
3.3. Detection of EGFR Protein Expression in Human and Murine TNBC Cell Lines
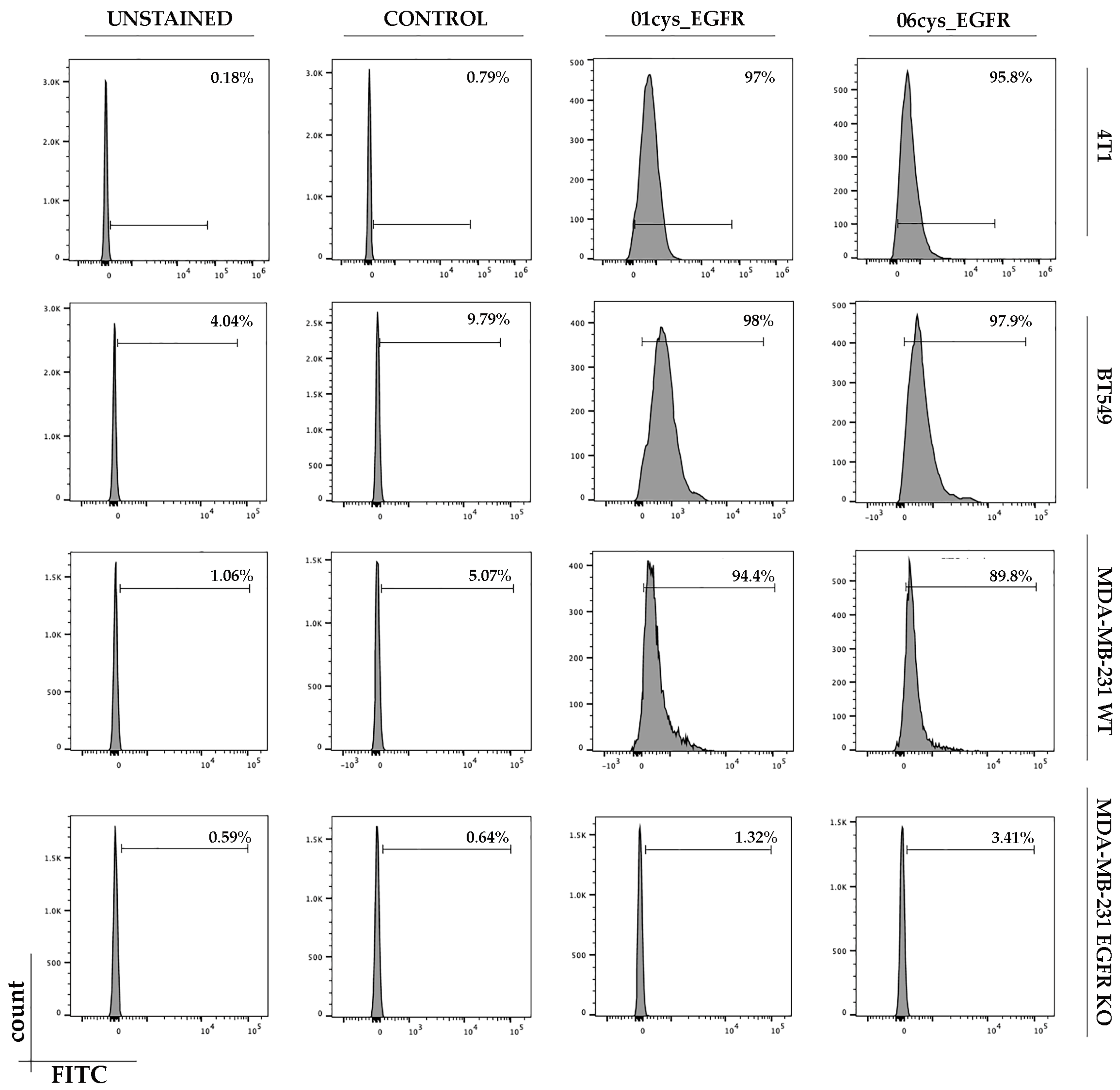
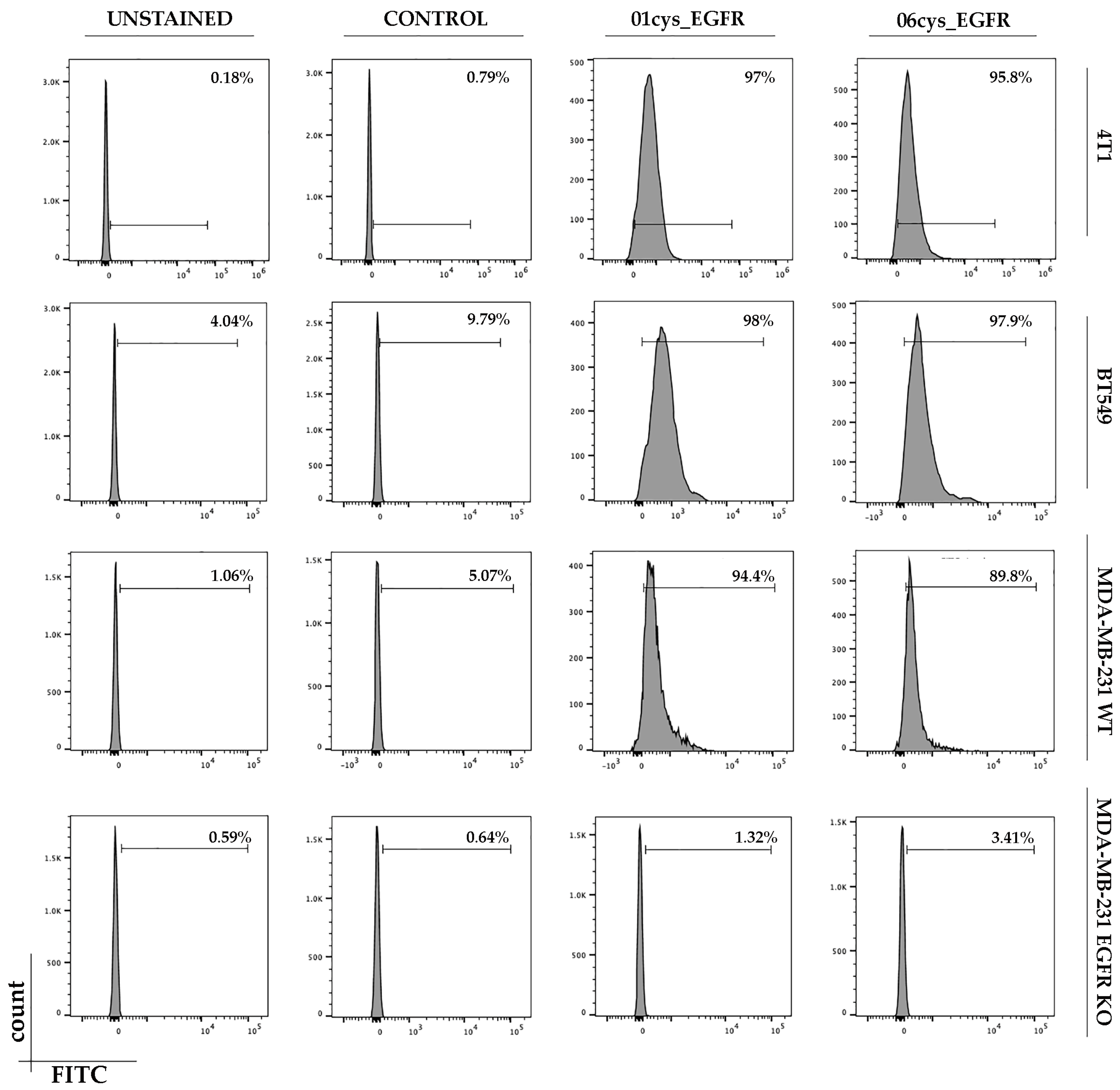
3.4. FITC-Conjugate Peptide Binding to Breast Cancer Cell Lines
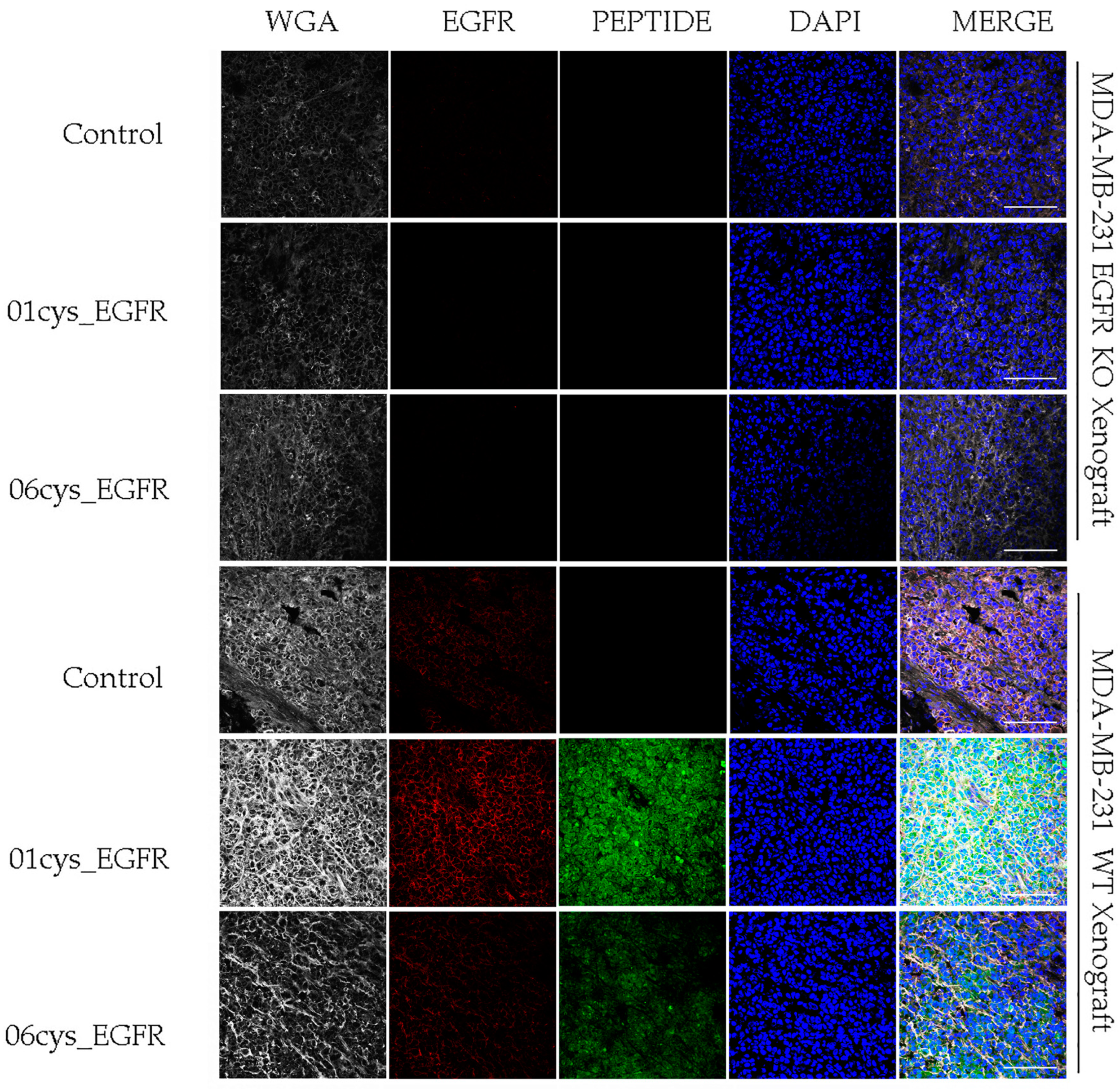
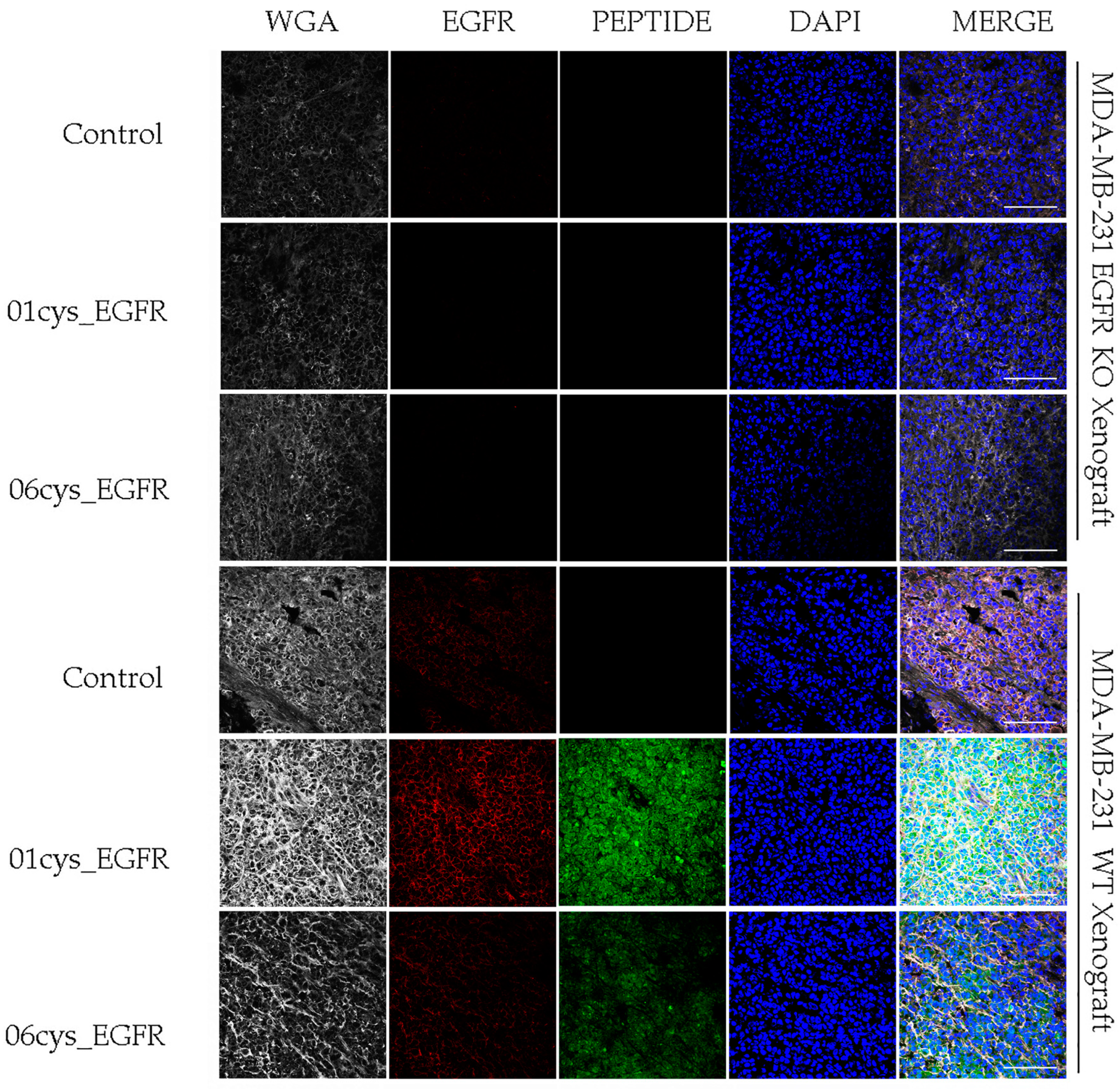
3.5. Ex Vivo EGFR-Specific Targeting of 01_cysEGFR and 06_cysEGFR Peptides
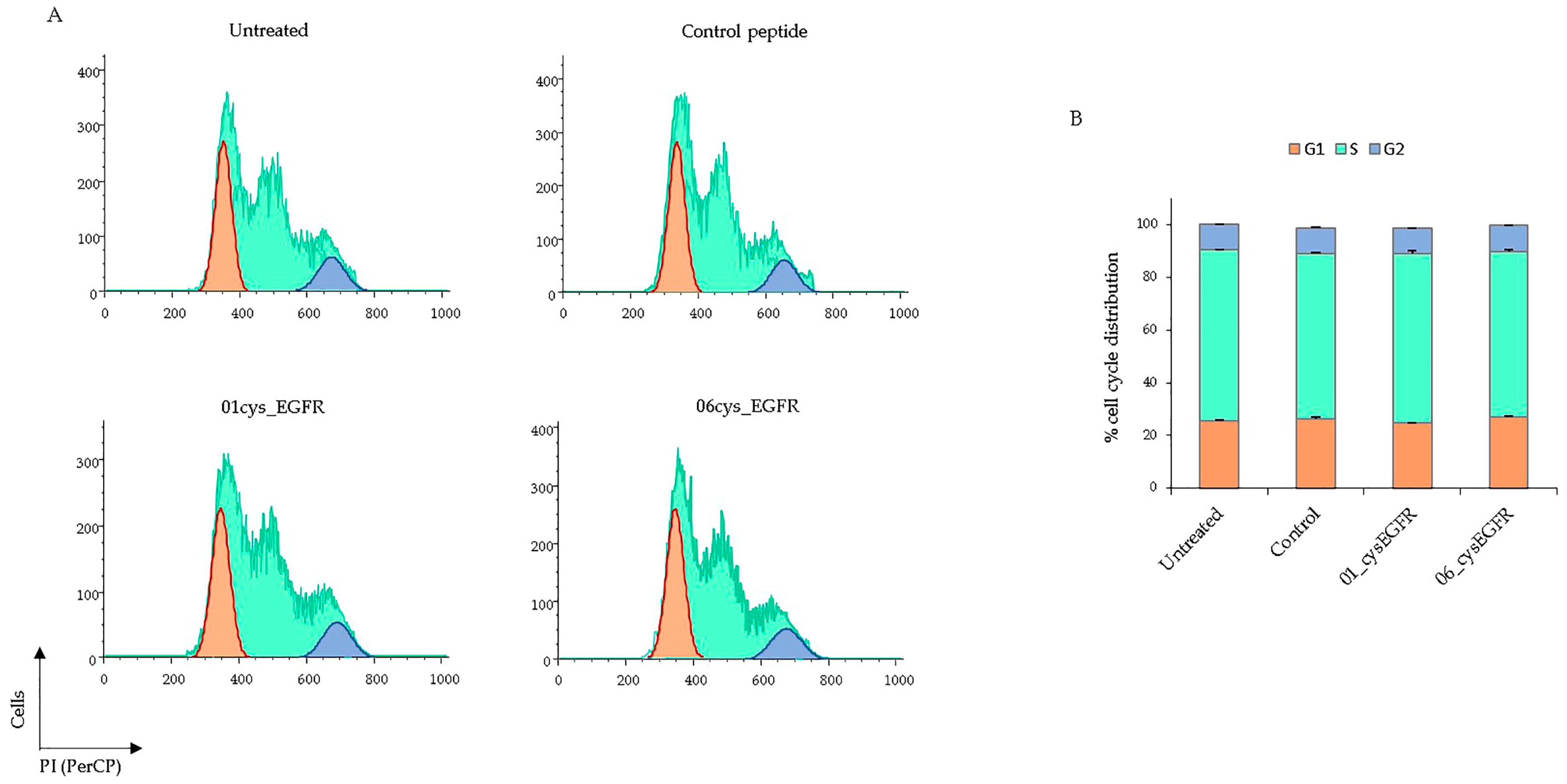
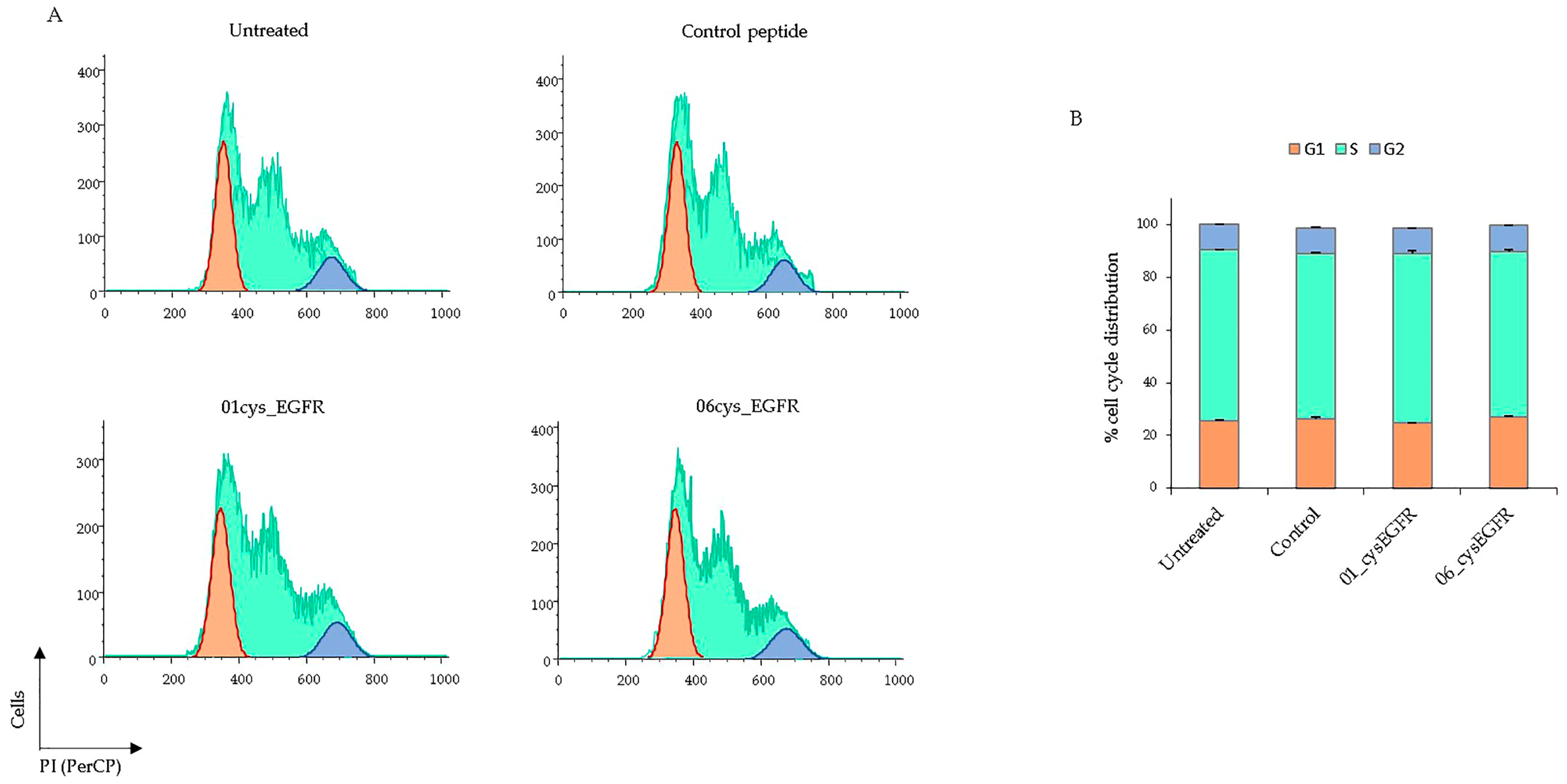
3.6. Peptides 01cys_EGFR and 06cys_EGFR Did Not Affect the Cell Cycle
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Viale, G.; Di Leo, A. Management of triple negative breast cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Penault-Llorca, F.; Viale, G. Phathological and molecular diagnosis of triple negative breast cancer: A clinical perspective. Ann. Oncol. 2012, 23, vi19–vi22. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Tan, K.; Rajan, R.S.; Mokhtar, N.; Adzmi, E.M.; Rahman, W.W.A.; Din, T.T.; Balakrishnan, V. Triple Negative Breast Cancer: A Review of Present and Future Diagnostic Modalities. Medicina 2021, 57, 62. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Van Reesema, L.L.S.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer: Who Should Receive Neoadjuvant Chemotherapy? Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, L.; Huang, B.; Li, X.; Yang, L.; Hu, X.; Jiang, Y.; Shao, Z.; Wang, Z. Efficacy and mechanism of the combination of PARP and CDK4/6 inhibitors in the treatment of triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 122. [Google Scholar] [CrossRef]
- Li, Y.; Zhan, Z.; Yin, X.; Fu, S.; Deng, X. Targeted Therapeutic Strategies for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 731535. [Google Scholar] [CrossRef]
- Hwang, S.-Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharm. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef]
- Bagegni, N.A.; Davis, A.A.; Clifton, K.K.; Ademuyiwa, F.O. Targeted Treatment for High-Risk Early-Stage Triple-Negative Breast Cancer: Spotlight on Pembrolizumab. Breast Cancer Targets Ther. 2022, 14, 113–123. [Google Scholar] [CrossRef]
- Lev, S. Targeted therapy and drug resistance in triple-negative breast cancer: The EGFR axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef]
- Klijn, J.G.M.; Berns, P.M.J.J.; Schmitz, P.I.M.; Foekens, J.A. The clinical significance of epidermal growth factor receptor (EGF-R) in human breast cancer: A review on 5232 patients. Endocr. Rev. 1992, 13, 3–17. [Google Scholar]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Naz, S.; Hashmi, S.K.; Irfan, M.; Hussain, Z.F.; Khan, E.Y.; Asif, H.; Faridi, N. Epidermal growth factor receptor (EGFR) overexpression in triple-negative breast cancer: Association with clinicopathologic features and prognostic parameters. Surg. Exp. Pathol. 2019, 2, 6. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Mi, L.-Z.; Schürpf, T.; Walz, T.; Springer, T.A. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Johnston, S.R.; Hickish, T.; Ellis, P.; Houston, S.; Kelland, L.; Dowsett, M.; Salter, J.; Michiels, B.; Ruixo, J.J.P.; Palmer, P.; et al. Phase II study of the efficacy and tolerability of two dosing regimens of the farnesyl transferase inhibitor, R115777, in advanced breast cancer. J. Clin. Oncol. 2003, 21, 2492–2499. [Google Scholar] [CrossRef]
- Cardoso, F.; Piccart, M.J.; Durbecq, V.; Di Leo, A. Resistance to trastuzumab: A necessary evil or a temporary challenge? Clin. Breast Cancer 2002, 3, 247–259. [Google Scholar] [CrossRef]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting Epidermal Growth Factor Receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef]
- Aloisio, A.; Nisticò, N.; Mimmi, S.; Maisano, D.; Vecchio, E.; Fiume, G.; Iaccino, E.; Quinto, I. Phage-Displayed Peptides for Targeting Tyrosine Kinase Membrane Receptors in Cancer Therapy. Viruses 2021, 13, 649. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Lowman, H.B. Phage Display for Protein Binding. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: New York, NY, USA, 2013; pp. 431–436. [Google Scholar]
- Lupia, A.; Mimmi, S.; Iaccino, E.; Maisano, D.; Moraca, F.; Talarico, C.; Vecchio, E.; Fiume, G.; Ortuso, F.; Scala, G.; et al. Molecular modelling of epitopes recognized by neoplastic B lymphocytes in Chronic Lymphocytic Leukemia. Eur. J. Med. Chem. 2020, 185, 111838. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J. Designing Peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef] [PubMed]
- Tomasella, C.; Floris, M.; Guccione, S.; Pappalardo, M.; Basile, L. Peptidomimetics in Silico. Mol. Inform. 2021, 40, 2000087. [Google Scholar] [CrossRef]
- Perez, J.J.; Perez, R.A.; Perez, A. Computational Modeling as a Tool to Investigate PPI: From Drug Design to Tissue Engineering. Front. Mol. Biosci. 2021, 8, 681617. [Google Scholar] [CrossRef]
- Hamzeh-Mivehroud, M.; Mahmoudpour, A.; Dastmalchi, S. Identification of new peptide ligands for epidermal growth factor receptor using phage display and computationally modeling their mode of binding. Chem. Biol. Drug Des. 2012, 79, 246–259. [Google Scholar] [CrossRef]
- Zhou, J.; Joshi, B.P.; Duan, X.; Pant, A.; Qiu, Z.; Kuick, R.; Owens, S.R.; Wang, T.D. EGFR Overexpressed in Colonic Neoplasia Can be Detected on Wide-Field Endoscopic Imaging. Clin. Transl. Gastroenterol. 2015, 6, e101. [Google Scholar] [CrossRef]
- Xue, E.Y.; Wong, R.C.H.; Wong, C.T.T.; Fong, W.-P.; Ng, D.K.P. Synthesis and biological evaluation of an epidermal growth factor receptor-targeted peptide-conjugated phthalocyanine-based photosensitiser. RSC Adv. 2019, 9, 20652–20662. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, W.; Cheng, L.; Meng, F.; Zhang, J.; Zhong, Z. EGFR-targeted multifunctional polymersomal doxorubicin induces selective and potent suppression of orthotopic human liver cancer in vivo. Acta Biomater. 2017, 64, 323–333. [Google Scholar] [CrossRef]
- Zou, Y.; Xia, Y.; Meng, F.; Zhang, J.; Zhong, Z. GE11-Directed Functional Polymersomal Doxorubicin as an Advanced Alternative to Clinical Liposomal Formulation for Ovarian Cancer Treatment. Mol. Pharm. 2018, 15, 3664–3671. [Google Scholar] [CrossRef]
- Li, C.; Cai, G.; Song, D.; Gao, R.; Teng, P.; Zhou, L.; Ji, Q.; Sui, H.; Cai, J.; Li, Q.; et al. Development of EGFR-targeted evodiamine nanoparticles for the treatment of colorectal cancer. Biomater. Sci. 2019, 7, 3627–3639. [Google Scholar] [CrossRef]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small Peptide Ligands for Targeting EGFR in Triple Negative Breast Cancer Cells. Sci. Rep. 2019, 9, 2723. [Google Scholar] [CrossRef] [Green Version]
- Mansour, S.; Adhya, I.; Lebleu, C.; Dumpati, R.; Rehan, A.; Chall, S.; Dai, J.; Errasti, G.; Delacroix, T.; Chakrabarti, R. Identification of a novel peptide ligand for the cancer-specific receptor mutation EGFRvIII using high-throughput sequencing of phage-selected peptides. Sci. Rep. 2022, 12, 20725. [Google Scholar] [CrossRef]
- Agnello, L.; Tortorella, S.; d’Argenio, A.; Carbone, C.; Camorani, S.; Locatelli, E.; Auletta, L.; Sorrentino, D.; Fedele, M.; Zannetti, A.; et al. Optimizing cisplatin delivery to triple-negative breast cancer through novel EGFR aptamer-conjugated polymeric nanovectors. J. Exp. Clin. Cancer Res. 2021, 40, 239. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Sviluppo e test del campo di forza tutto atomico OPLS sull’energetica conformazionale e le proprietà dei liquidi organici. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassenaar, T.A.; van Dijk, M.; Loureiro-Ferreira, N.; Van Der Schot, G.; De Vries, S.J.; Schmitz, C.; Van Der Zwan, J.; Boelens, R.; Giachetti, A.; Ferella, L.; et al. WeNMR: Structural Biology on the Grid. J. Grid Comput. 2012, 10, 743–767. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein-protein complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Release, S. Maestro, Version 11.5.011, Schrödinger, LLC, NY 2018–1. Available online: https://www.schrodinger.com/products/maestro. (accessed on 3 May 2021).
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.Z.; Grey, M.J.; Zhu, J.; Graef, E.; Yokoyama, S.; Springer, T.A. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell Biol. 2010, 30, 5432–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.E.M.; de Vries, S.J.; Zacharias, M. Fully Blind Peptide-Protein Docking with pepATTRACT. Structure 2015, 23, 1507–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [Green Version]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved docking of polypeptides with Glide. J. Chem. Inf. Model. 2013, 53, 1689–1699. [Google Scholar] [CrossRef]
- Antes, I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins 2010, 78, 1084–1104. [Google Scholar] [CrossRef]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef]
- Sanders, J.M.; Wampole, M.E.; Thakur, M.L.; Wickstrom, E. Molecular determinants of epidermal growth factor binding: A molecular dynamics study. PLoS ONE 2013, 8, e54136. [Google Scholar] [CrossRef] [Green Version]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HADDOCK Score * | Cluster Size | RMSD ** | VdW Evdw | Electrostatic Eelec | Desolvation Energy | Buried Surface Area | Z-Score *** | |
|---|---|---|---|---|---|---|---|---|
| 01_cysEGFR | −42.4 ± 5.7 | 86 | 0.4 ± 0.3 | −37.5 ± 2.2 | −111.7 ± 22.3 | 0.8 ± 2.2 | 1176.4 ± 56.0 | −1.6 |
| 06_cys EGFR | −47.4 ± 1.3 | 28 | 1.6 ± 0.1 | −33.2 ± 3.5 | −198.4 ± 42.3 | 6.4 ± 0.6 | 1097.1 ± 37.4 | −1.8 |
| Binding Affinity calculated with PRODIGY server | ||||||||
| Binding Affinity (ΔG) § | Predicted Dissociation Constant (KD) | Temperature (°C) | ||||||
| 01_cysEGFR | −10.1 | 7.7 × 10−8 | 37 | |||||
| 06_cysEGFR | −8.5 | 1.1 × 10−6 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisticò, N.; Aloisio, A.; Lupia, A.; Zimbo, A.M.; Mimmi, S.; Maisano, D.; Russo, R.; Marino, F.; Scalise, M.; Chiarella, E.; et al. Development of Cyclic Peptides Targeting the Epidermal Growth Factor Receptor in Mesenchymal Triple-Negative Breast Cancer Subtype. Cells 2023, 12, 1078. https://doi.org/10.3390/cells12071078
Nisticò N, Aloisio A, Lupia A, Zimbo AM, Mimmi S, Maisano D, Russo R, Marino F, Scalise M, Chiarella E, et al. Development of Cyclic Peptides Targeting the Epidermal Growth Factor Receptor in Mesenchymal Triple-Negative Breast Cancer Subtype. Cells. 2023; 12(7):1078. https://doi.org/10.3390/cells12071078
Chicago/Turabian StyleNisticò, Nancy, Annamaria Aloisio, Antonio Lupia, Anna Maria Zimbo, Selena Mimmi, Domenico Maisano, Rossella Russo, Fabiola Marino, Mariangela Scalise, Emanuela Chiarella, and et al. 2023. "Development of Cyclic Peptides Targeting the Epidermal Growth Factor Receptor in Mesenchymal Triple-Negative Breast Cancer Subtype" Cells 12, no. 7: 1078. https://doi.org/10.3390/cells12071078
APA StyleNisticò, N., Aloisio, A., Lupia, A., Zimbo, A. M., Mimmi, S., Maisano, D., Russo, R., Marino, F., Scalise, M., Chiarella, E., Mancuso, T., Fiume, G., Omodei, D., Zannetti, A., Salvatore, G., Quinto, I., & Iaccino, E. (2023). Development of Cyclic Peptides Targeting the Epidermal Growth Factor Receptor in Mesenchymal Triple-Negative Breast Cancer Subtype. Cells, 12(7), 1078. https://doi.org/10.3390/cells12071078